
INHIBICJA PROTEASOMĂ“W JAKO NOWY CEL TERAPEUTYCZNY W CHOROBACH NOWOTWOROWYCH
“Proteasome inhibition as a novel therapeutic target in neoplasmatic diseases”
Streszczenie:
Proteasom 26S jest dużą, wewnątrzkomórkową proteazą, rozpoznającą i degradującą białka zaznaczone przez układ ubikwityny do zniszczenia. Właściwa degradacja białek komórkowych ma znaczenie dla czynności komórek prawidłowych, a inhibicja szlaku proteasomów prowadzi do zatrzymania cyklu komórkowego i apoptozy. Zaburzenia regulacji tego układu enzymatycznego mogą również odgrywać rolę w progresji nowotworu i oporności na leki, co czyni inhibicję proteasomów nowym celem terapeutycznym. Bortezomib (PS-341) jest pierwszym inhibitorem proteasomów, który wszedł do praktyki klinicznej. Jest to dipeptyd kwasu boronowego, bezpośrednio inhibujący kompleks enzymatyczny. Ostatnio wykazano aktywność bortezomibu w kilku typach nowotworów, co potwierdza wartość terapeutyczną hamowania proteasomów w tych chorobach u człowieka. Został on zatwierdzony w 2003 roku do leczenia opornego szpiczaka mnogiego (MM), a blisko jedna trzecia pacjentów z nawrotem MM wykazywała istotne korzyści kliniczne w dużych badaniach klinicznych. Mechanizm jego działania jest częściowo mediowany przez inhibicję czynnika jądrowego-kappa B, prowadząc do apoptozy, zmniejszenia ekspresji cytokin angiogennych i hamowania przylegania komórek nowotworowych do zrębu. Obecnie trwa kilka badań klinicznych w MM, jak i w innych nowotworach złośliwych. Niniejszy artykuł omawia inhibicję proteasomów jako nowy cel terapeutyczny oraz skupia się na mechanizmach działania i aktualnych doświadczeniach klinicznych z bortezomibem.
Abstrakt:
The 26S proteasome is a large intracellular protease that identifies and degrades proteins tagged for destruction by the ubiquitin system. The orderly degradation of cellular proteins is critical for normal cell cycling and function, and inhibition of the proteasome pathway results in cell-cycle arrest and apoptosis. Dysregulation of this enzymatic system may also play a role in tumor progression, drug resistance, and altered immune surveillance, making the proteasome an appropriate and novel therapeutic target in cancer. Bortezomib (PS-341) is the first proteasome inhibitor to enter clinical practice. It is a boronic aid dipeptide that binds directly with and inhibits the enzymatic complex. Bortezomib has recently shown significant preclinical and clinical activity in several cancers, confirming the therapeutic value of proteasome inhibition in human malignancy. It was approved in 2003 for the treatment of advanced multiple myeloma (MM), with approximately one third of patients with relapsed and refractory MM showing significant clinical benefit in a large clinical trial. Its mechanism of action is partly mediated through nuclear factor-kappa B inhibition, resulting in apoptosis, decreased angiogenic cytokine expression, and inhibition of tumor cell adhesion to stroma. Several clinical trials are currently ongoing in MM as well as several other malignancies. This article discusses proteasome inhibition as a novel therapeutic target in cancer and focuses on the development, mechanism of action, and current clinical experience with bortezomib.
Wprowadzenie
Wielokatalityczny szlak ubikwityna-proteasomy odpowiada za degradację białek komórkowych.1 Proces ten jest zależny od adenozyno-5’-trójfosforanu i jest właściwy dla prawidłowych cykli komórkowych, co czyni inhibicję proteasomów nowym celem terapeutycznym w komórkach nowotworowych. Bortezomib jest pierwszym inhibitorem szlaku proteasomów, który wszedł do praktyki klinicznej.1,2 W dużym, wieloośrodkowym badaniu klinicznym fazy II, blisko jedna trzecia pacjentów z zaawansowanym szpiczakiem mnogim (MM) wykazywała istotną odpowiedź na leczenie bortezomibem.3 Na podstawie tych wyników w dniu 13 maja 2003 roku, Urząd ds. Żywności i Rejestracji Leków Stanów Zjednoczonych (FDA) udzielił przyspieszonego zezwolenia dla stosowania tego leku u chorych z MM, którzy mieli nawrót, po co najmniej dwóch schematach leczenia i stwierdzono u nich dowody oporności na ostatnio stosowane u nich leczenie. Wysoce obiecująca aktywność kliniczna wykazywana przez bortezomib w MM i innych nowotworach złośliwych potwierdziła, że proteasomy są nowym, ważnym celem terapetycznym.
Szlak ubikwityna-proteasomy
Sprawna degradacja białek komórkowych przez szlak ubikwityna-proteasomy jest istotna dla transdukcji sygnału, regulacji transkrypcji, odpowiedzi na stres oraz kontroli czynności receptorów.4 Szlak ten kontroluje aktywację czynnika jądrowego-kappa B (NF-?B, ważny czynnik transkrypcyjny) poprzez degradację inhibitora NF-?B (I-?B).5 Proteasomy odgrywają również ważną rolę w prezentacji antygenu. W nowotworze, dysregulacja tego procesu katalitycznego może przyczyniać się do progresji nowotworu, oporności na leki i zmian w kontrolnych mechanizmach immunologicznych.6 Zatem jakakolwiek inhibicja układu proteasomów zaburza równowagę pomiędzy białkami regulatorowymi, co wywołuje zatrzymanie cyklu komórkowego w fazach G1-S i G2-M cyklu komórkowego oraz apoptozę.
Pierwszym krokiem w szlaku ubikwityna-proteasomy jest dołączenie łańcuchów poliubikwitynowych do białek przeznaczonych do zniszczenia (Rycina 1). Ubikwityna jest małym białkiem o zdolności tworzenia łańcuchów multimerycznych. Proces degradacji jest ściśle regulowany, a swoiste białka mogą być ukierunkowywane poprzez kontrolowanie powinowactwa ubikwityny do danego substratu.18 Drugim krokiem w procesie katalitycznym jest identyfikacja ubikwitynowanych białek przez wewnątrzkomórkowy kompleks proteasomowy. Końcowym etapem jest degradacja wyznaczonych białek w centralnej części kompleksu proteasomowego.
Budowa kompleksu proteasomowego
Proteasom 26 S (1500–2000 kd) składa się z rdzeniowego kompleksu katalitycznego 20S (około 700 kd) i kompleksu regulatorowego 19S (Rycina 1).1,4,7 Składa się on z dwóch zewnętrznych i dwóch wewnętrznych pierścieni, które są ułożone tak, że formują strukturę cylindryczną posiadającą trzy przedziały.8 Każdy pierścień zewnętrzny posiada siedem podjednostek alfa (?1–?7), podczas gdy każdy pierścień wewnętrzny zawiera siedem podjednostek beta (?1–?7). Kompleks proteasomowy 20S posiada aktywności chymotrypsyno-, trypsyno- i kaspazopodobne.7 Konformacyjnie jest elastyczny; aktywne miejsca katalityczne umiejscowione są na wewnętrznej powierzchni cylindra, gdzie wiążą się substraty białkowe.9
Białka znakowane ubikwityną są rozpoznawane przez kompleks regulatorowy 19S, gdzie też następuje usuwanie znaczników ubikwitynowych. ATP-azy o aktywności chaperono-podobnej u podstawy kompleksu regulatorowego 19S rozwijają następnie substraty białkowe i wprowadzają je wewnętrznych przedziałów katalitycznych cylindra proteasomowego 20S.10 Otwór wejściowy do komory katalitycznej 20S jest mały (około 1,3 nm) i konieczne jest znaczne rozwinięcie substratu.9 Otworu wejściowego strzeże także bramka molekularna (N-końcowy fragment łańcucha podjednostki ?3), ale jest ona konstytutywnie otwarta, kiedy podjednostki regulatorowe 19S są związane z proteasomem 20S.11 Białka wchodzące do komory wewnętrznej ulegają hydrolizie przez sześć aktywnych miejsc proteolitycznych w podjednostkach ? (dwa miejsca na każdej podjednostce ?1, ?2 i ?5) na małe polipeptydy o zakresie długości 3–22 aminokwasów.1,8 Białka nie mogą wchodzić do wewnętrznego cylindra przez zewnętrzne ściany proteasomu 20S, ponieważ szczeliny pomiędzy pierścieniami są wąskie.
Inhibitory proteasomĂłw
Degradacji przez szlak proteasomów ulega kilka białek regulatorowych, supresorów nowotworowych, czynników transkrypcyjnych i onkogenów (Tabela 1). Inhibicja proteasomów może wywoływać apoptozę przez wpływ na stężenia różnych białek, co prowadzi do inhibicji aktywność NF-?B, zwiększenia aktywności białek p53 i Bax oraz akumulacji inhibitorów p27 i p21 kinazy zależnej od cyklin.7 Badania przedkliniczne wykazują, że komórki nowotworów złośliwych, transformowane i proliferujące są bardziej podatne na inhibicje proteasomów niż komórki prawidłowe.1,29–32 Stworzono i opisano liczne inhibitory proteasomów.4,7,13 Imajoh-Ohmi i wsp.14 wykazali, że laktacystyna (nieodwracalny inhibitor podjednostki katalitycznej ? proteasomu) indukuje apoptozę ludzkich komórek monoblastycznych U937. Następnie Shinohara i wsp.15 wykazali, że benzylooksykarbonylo-(Z)-Leu-Leu-leucynal (tripeptydowo-aldehydowy inhibitor proteasomów) indukuje w komórkach białaczkowych apoptozę zależną od p53.
Powyższe oraz inne badania dostarczyły zasadniczych dowodów na to, że proteasomy są ważnym celem leczenia przeciwnowotworowego, jednakże dostępnym inhibitorom brakuje swoistości.4,7 Dlatego też Adams i wsp.16 zaprojektowali i stworzyli kilka związków będących pochodnymi kwasu boronowego, hamujących szlak proteasomów w sposób wysoce swoisty. Większość z tych inhibitorów proteasomów wykazywało aktywność w obrębie panelu 60 nowotworowych linii komórkowych z Narodowego Instytutu Raka (NCI), a siła inhibicji proteasomów korelowała z działaniami hamującymi wzrost. Na podstawie swojej mocy i cytotoksyczności, bortezomib został zidentyfikowany jako najlepszy kandydat do dalszych badań klinicznych.
Bortezomib: aktywność przedkliniczna
Bortezomib (kwas N-pyrazynokarbonylo-L-fenyloalanino-L-leucyno-boronowy; znany uprzednio jako PS-341 lub MLN-341), dipeptyd kwasu boronowego, jest unikalnym i swoistym inhibitorem szlaku proteasomów.3,16 Boretzomib inhibuje szlak proteasomów szybko i w sposób odwracalny, poprzez wiązanie się bezpośrednio do kompleksu proteasomowego 20S i hamowanie jego aktywności enzymatycznej. W modelach zwierzęcych, bortezomib nie przechodzi do mózgu, rdzenia kręgowego, jąder lub oczu, oszczędzając w ten sposób te tkanki od efektów ubocznych inhibicji proteasomów. W związku z tym, że nie obserwowano inhibicji proteasomów w tkankach nowotworowych izolowanych z mózgu i jąder, jest jednakże mało prawdopodobne, że inhibitory proteasomów będą użyteczne w tych typach nowotworów, chyba, że bariera krew-mózg lub krew jądra zostanie przerwana przez proces nowotworowy.16
Badania przedkliniczne wykazują, że cytotoksyczne i hamujące wzrost działania bortezomibu korelują z inhibicją proteasomów, niezależnie od stanu p53 i nie nakładają się z innymi lekami chemioterapeutycznymi.16
Badania kliniczne fazy I z bortezomibem
Na podstawie obiecujących badań przedklinicznych opisanych wcześniej, Orlowski i wsp.17 przeprowadzili badanie fazy I, w którym 27 pacjentów z zaawansowanymi, opornymi na leczenie hematologicznymi nowotworami złośliwymi było leczonych bortezomibem. Maksymalna dawka tolerowana wynosiła 1,04 mg/m2, podawana w dni 1, 4, 8, 11, 15, 18, 22 i 25, z następowymi 2 tygodniami okresu odpoczynku (cykl 6-tygodniowy). Kluczowym wynikiem tego badania było to, że obserwowano odpowiedzi u dziewięciu pacjentów z MM leczonych w tym badaniu, w tym u jednego pacjenta z opornym na leczenie nawrotem MM, który osiągnął odpowiedź całkowitą (CR). W badaniu fazy I stwierdzono dwie inne odpowiedzi – jedną obserwowano u chorego z chłoniakiem z komórek strefy płaszcza, a druga wystąpiła u pacjenta z chłoniakiem folikularnym. W innym badaniu fazy I, bortezomib badano u 43 pacjentów z zaawansowanymi nowotworami litymi.18 Głównymi, ograniczającymi dawkę efektami ubocznymi były biegunka i neuropatia. Schemat dawkowania bardzo zbliżony to stosowanego w badaniach nad MM, mianowicie leczenie podawano dwa razy w tygodniu przez 2 tygodnie (w dni 1, 4, 8 i 11) z następowym 1 tygodniem odpoczynku. Maksymalna dawka tolerowalna w tym badaniu wynosiła 1,56 mg/m2.
Racjonalne uzasadnienie badań klinicznych nad leczeniem bortezomibem w MM
Dodatkowo do wydajności klinicznej wykazanej w warunkach fazy I, bortezomib wykazywał uderzającą aktywność przeciwko komórkom MM w kilku modelach przedklinicznych. Bezpośrednio hamował proliferację komórek MM, indukował apoptozę w liniach komórkowych i pierwotnych komórkach MM oraz inhibował wiązanie patologicznych plazmocytów do komórek zrębowych szpiku kostnego.19 Deksametazon nasilał aktywność bortezomibu, a interleukina-6 (IL-6) nie chroniła komórek MM przed apoptozą. Bortezomib indukował nieodwracalna apoptozę w komórkach z zarówno dzikim typem p53, jak i zmutowaną wersją p53. Uwrażliwiał również linie komórek MM i komórki pacjentów na inne substancje czynne, takie jak doksorubicyna i melfalan.20 Następnie zbadano bortezomib w mysim modelu ksenogenicznego przeszczepu ludzkiego szpiczaka.21 U myszy leczonych bortezomibem stwierdzono istotną inhibicję wzrostu, w tym kilka całkowitych regresji nowotworu. Przeżycie całkowite było u leczonych myszy dwukrotnie dłuższe niż w grupie kontrolnej, a leczenie wiązało się z apoptozą komórek nowotworowych i zmniejszeniem angiogenezy.21 Te wyniki przedkliniczne, łącznie z dowodami aktywności w badaniach fazy I, dostarczyły racjonalnego podłoża dla badań klinicznych fazy II nad bortezomibem w MM.
Badania kliniczne nad bortezomibem w MM
W niedawnym, wieloośrodkowym badaniu fazy II nad bortezomibem w MM, leczono 202 dwóch pacjentów z opornym na leczenie nawrotem MM.3 Wymagano by pacjenci mieli mierzalne stężenia białka monoklonalnego i byli oporni na leczenie ratujące, co definiowano jako chorobę postępującą przy leczeniu lub progresję choroby w ciągu 60 dni od ukończenia leczenia. Bortezomib podawano w dawce początkowej 1,3 mg/m2 i.v. przez 3–5 sekund dwa razy w tygodniu przez 2 tygodnie w dni 1, 4, 8 i 11, z pozwoleniem na zmniejszenia dawki przy toksyczności. Leczenie powtarzano co 21 dni przez maksymalnie 8 cykli. U pacjentów z chorobą postępującą po dwóch cyklach lub chorobą stabilną po czterech cyklach dołączano deksametazon. Dawka deksometazonu obejmowała 20 mg w dniu podawania każdej dawki boretzomibu i w dniu następnym. Ocenie poddano stu dziewięćdziesięciu trzech pacjentów. U większości z nich (n = 178) nie powiodło się leczenie wszystkimi znanymi stosowanymi w MM aktywnymi klasami leków, przy medianie liczby terapii uprzednich terapii wynoszącej sześć (w zakresie 2–15). Spośród poddanych ocenie pacjentów, 53 (27%) osiągnęło częściową odpowiedź na leczenie, zdefiniowaną według kryteriów European Group for Blood and Marrow Transplantation.22 Ponadto, 14 pacjentów (7%) osiągnęło minimalną odpowiedź na leczenie. Odpowiedzi obejmowały 4% pacjentów, którzy osiągnęli CR (z ujemną immunofiksacją), i dalsze 6% pacjentów z prawie-CR, którzy spełniali kryteria dla CR, ale u których utrzymywała się dodatnia immunofiksacja. Mediana czasu przeżycia dla wszystkich pacjentów wynosiła 16 miesięcy, a mediana czasu do progresji 7 miesięcy. Odpowiedzi były trwałe, z medianą czasu trwania odpowiedzi 12 miesięcy u chorych, którzy osiągnęli CR, odpowiedź częściową lub odpowiedź minimalną na leczenie. Odpowiedzi wiązały się z poprawą wartości hemoglobiny, liczby płytek, czynności nerek, stanu wydolności i wskaźników jakości życia.23 Najpowszechniejsze działania niepożądane obejmowały efekty uboczne żołądkowo-jelitowe, cytopenie (szczególnie trombocytopenię), zmęczenie i neuropatię obwodową. Wyniki te były szczególnie interesujące, biorąc pod uwagę, że u pacjentów stosowano uprzednio silne leczenie; ściśle rzecz ujmując u 64% z nich zastosowano uprzednio przeszczep komórek pnia, a u 83% stosowano uprzednio leczenie talidomiedem. Starszy wiek (>65 lat), obecność nieprawidłowości cytogenetycznych, i ponad 50%-owe zajęcie szpiku kostnego przez komórki plazmatyczne wiązały się z mniejszym odsetkiem odpowiedzi.3 Przeciwnie, delecja w obrębie chromosomu 13, wcześniejsze leczenie talidomiedem, i uprzedni przeszczep nie wpływały na odpowiedź na bortezomib.
Osobne badanie fazy II z randomizacją przeprowadzono u chorych, u których nie osiągnięto odpowiedzi lub mieli nawrót po terapii pierwszego rzutu w MM.24 W badaniu tym stosowano dwie następujące dawki bortezomibu u 54 pacjentów: 1,0 mg/m2 (28 pacjentów) w porównaniu do 1,3 mg/m2 (26 pacjentów) podawane w dni 1, 4, 8 i 11 co 21 dni. Odpowiedzi (CR, odpowiedź częściowa lub odpowiedź minimalna) obserwowano u 33% pacjentów przy dawce 1,0 mg/m2 i u 50% pacjentów przy dawce 1,3 mg/m2, co wydawało się sugerować zależność dawka-odpowiedź, chociaż autorzy nie przedstawili formalnego porównania statystycznego.
Chociaż obydwa badania fazy II wymagały maksymalnie 8 cykli terapii, to u pacjentów odpowiadających dozwolono podawanie dalszej kontynuacji leczenia w trzecim badaniu (protokół przedłużony). Dane odnośnie 57 pacjentów leczonych w tym przedłużonym badaniu wykazują, że bezpieczne jest podanie, co najmniej pięciu do sześciu dodatkowych cykli terapii, przy podobnym profilu toksyczności jak w pierwszych ośmiu cyklach.24
Zalecenia dla stosowania bortezomibu w MM
Pacjentów z nawrotem MM leczy się zazwyczaj konwencjonalną chemioterapią, dużymi dawkami kortykosteroidów lub talidomidem. U tych chorych, bortezomib stanowi obecnie ważną opcję dodatkową. Ponieważ żaden z terapeutycznych sposobów podejścia nie ulecza, chorych zazwyczaj leczy się stosując wymienione opcje sekwencyjnie, a wybór terapii przy każdym nawrocie powinno się określać na podstawie sytuacji klinicznej i preferencji pacjenta. Chorzy, którzy nie odpowiadają na leczenie indukcyjne MM często odnoszą korzyść z autologicznego przeszczepu komórek pnia ponieważ intensywność dawki leczenia kondycjonującego opartego na melfalanie pokonuje lekooporność.25 Bortezomib może się okazać wartościowy i może ułatwiać osiąganie CR po przeszczepie, kluczowego celu terapeutycznego w MM. Wstępne dane wykazują, że w połączeniu z deksametazonem, bortezomib ma imponującą aktywność jako leczenie przedtransplantacyjne w MM, z odsetkami odpowiedzi przekraczającymi 75%.26 Podczas „XI International Myeloma Workshop”, które odbyły się w Sydney w kwietniu 2005 roku doniesiono o bardzo obiecujących wstępnych próbach łączenia, Bortezomibu oraz CC-5013, osiągając odpowiedź pozytywną u ponad 90% pacjentów z MM.
Aktywność bortezomibu w innych nowotworach złośliwych
Aktualnie trwa klika badań klinicznych z bortezomibem w różnych hematologicznych i innych nowotworach złośliwych. Dane relacjonowane jak dotychczas z badań fazy II nad bortezomibem przedstawiono w Tabeli 2.3,24,27-32 Wstępne wyniki sugerują obiecującą aktywność w chłoniakach z komórek strefy płaszcza i być może w chłoniakach folikularnych oraz limfocytarnych z małych limfocytów o niskim stopniu zaawansowania.33
Mechanizm działania bortezomibu
Przeciwnowotworowe działania bortezomibu są wynikiem apoptozy komórkowego zatrzymania cyklu komórkowego, które wynikają z efektów inhibicji proteasomów (Tabela 1). Leżące u podłoża mechanizmy obejmują inhibicję NF-?B, zwiększenie aktywności szlaków apoptotycznych i działania na mikrośrodowisko nowotworu. Te konsekwencje inhibicji proteasomów zbadano dobrze w kontekście MM, gdzie staje się to aktualnie coraz bardziej oczywiste, że zarówno komórki nowotworu złośliwego, jak i jego mikrośrodowisko są ważnymi celami terapii.34 Nowe leki, takie jak talidomid i jego analog CC-5013 są reprezentatywne dla tej nowej strategii terapeutycznej w MM,35 a aktualne dane popierają podobną podwójną rolę bortezomibu (Rycina 2).20,36
Inhibicja NF-?B
Kilka efektów bortezomibu, w tym apoptoza, wydaje się być mediowane poprzez inhibicję NF-?B.37 Rodzina białek Rel/NF-?B to indukowalne dimeryczne czynniki transkrypcyjne rozpoznające i wiążące wspólny motyw sekwencji w jądrowym DNA.37 NF-?B, główny czynnik transkrypcyjny z tej rodziny, jest heterodimerem p50/RelA (p50/p65) obecnym w cytoplazmie prawie wszystkich komórek.38 NF-?B reguluje wzrost komórek i apoptozę, jak również ekspresje różnych cytokin, cząsteczek adhezyjnych i ich receptorów.7 W cytoplazmie, NF-?B jest normalnie połączony ze swoim inhibitorem – I-?B. Gdy komórki ulegają stymulacji (przez cytokiny, stres lub chemioterapię) aktywacji ulegają kaskady sygnałowe prowadzące do aktywacji kinazy I-?B, heterodimerycznej kinazy białkowej katalizującej fosforylację I-?B (Rycina 3). Kinaza I-?B fosforyluje dwie reszty serynowe w N-końcowej domenie regulatorowej I-?B.39 Ufosforylowane miejsca w I-?B są wtedy rozpoznawane przez E3RS (I-?B/?-TrCP), ligazę ubikwityny E3 typu SCF, co prowadzi do ubikwitynacji. I-?B jest następnie degradowane poprzez szlak proteasomów, uwalniając wolny, czynny NF-?B. Po aktywacji NF-?B przemieszcza się do jądra i wiąże do regionów promotorowych kilku genów docelowych, wywołując tym samym ich transkrypcję. Prowadzi to do zwiększenia ekspresji różnych cytokin i chemokin, cząsteczek adhezyjnych i cykliny D, sprzyjających wzrostowi i przeżyciu komórek.37
Inhibitory proteasomów hamują aktywność NF-?B w komórkach poprzez blokowanie degradacji I-?B.37 Inhibicja aktywności transkrypcyjnej NF-?B odgrywa korzystną rolę w nowotworach poprzez zmniejszenie ekspresji różnych czynników wzrostu, przeżycia i angiogenetycznych. Prowadzi to do zmniejszenia stężeń białek proapoptotycznych Bcl-2 i A1/Bfl-1, wywołujących uwalnianie cytochormu C, aktywację kaspazy-9 i apoptozę.20 Biorąc pod uwagę znaną rolę NF-?B w MM, inhibicja NF-?B jest prawdopodobnie jednym z głównych mechanizmów, na drodze, których bortezomib indukuje apoptozę i pokonuje lekooporność.37
NF-?B jest również ważny dla ekspresji komórkowych cząsteczek adhezyjnych. W MM, aktywacja NF-?B prowadzi do zwiększenia ekspresji przez komórki plazmatyczne cząsteczek adhezyjnych, takich jak ICAM-1 i VCAM-1. Z kolei wiązanie komórek MM do zrębu wywołuje mediowane przez NF-?B zwiększenie wydzielania IL-6 przez komórki zrębu, co nadaje oporność na apoptozę i chemioterapię.40 Bortezomib inhibuje przyleganie komórek MM do zrębu, co częściowo wyjaśnia zahamowanie przez niego NF-?B.19
Aktywacja NF-?B promuje ekspresję różnych cytokin mediujących angiogenezę i wzrost. W mysim modelu MM, leczenie bortezomibem inhibuje angiogenezę, który to efekt również może się wiązać z inhibicją NF-?B.21 Ponadto bortezomib blokuje zależną od NF-?B indukcję wydzielania przez komórki zrębowe czynników wzrostu, takich jak IL-6.46
Chociaż, jak to wcześniej omówiono, inhibicja NF-?B może być głównym mechanizmem działania, to bortezomib wywiera prawdopodobnie także inne efekty, mające udział w jego aktywności przeciwnowotworowej, szczególnie w MM. PS-1145, swoisty inhibitor kinazy kinazy I-?B, wywołuje jedynie 20–50 procentową (%) inhibcję proliferacji komórek MM przy stężeniu większym niż 12,5 ?mol/L, w porównaniu z całkowitą inhibicją przy stosowaniu bortezomibu w stężeniu ? 0,1 ?mol/L.37 Co więcej, w odróżnieniu od PS-1145, bortezomib indukuje również apoptozę komórek MM.
Zwiększenie aktywności szlaków proapoptotycznych
Badania z wykorzystaniem technologii mikromacierzy w komórkach MM leczonych bortezomibem wykazują zwiększenie ekspresji genów białek szoku termicznego i genów proapoptotycznych, natomiast geny wzrostu i antyapoptotyczne ulegają supresji.20 Bortezomib aktywuje kinazę N-końcową c-Jun (JNK) prowadząc do zwiększenia ekspresji Fas i aktywacji kaspazy-8 i kaspazy-3.20 Ten mediowany przez kaspazę-8 szlak apoptotyczny jest niezależny od szlaku mediowanego przez kaspazę-9, opisanego wcześniej w odniesieniu do inhibicji NF-?B. Aktywacja JNK wydaje się być ważnym szlakiem dla indukowanej przez bortezomib apoptozy komórek MM, a blokada JNK przez swoisty inhibitor (SP600125) może hamować ten efekt poprzez blok aktywacji kaspazy-3. Indukcja kaspazy-3 prowadzi do degradacji MDM2 i fosforylacji p53 (ser 15), zwiększając przez to aktywność p53 i apoptozę. Bortezomib indukuje także ekspresję FasL, prawdopodobnie z powodu zwiększonej ekspresji c-myc, występującej w wyniku inhibicji proteasomów.20
Inne efekty
Bortezomib zmniejsza ekspresję insulino-podobnego czynnika wzrostu-1 i receptora insulino-podobnego czynnika wzrostu-1.20 Hamuje również indukowaną przez IL-6 aktywację szlaku Ras/Raf/kinaza białkowa aktywowana przez mitogeny, co prowadzi do inhibicji wzrostu w liniach komórkowych MM i pierwotnych komórkach MM.37 Bortezomib nie wywiera jednak żadnego działania na indukowaną przez IL-6 sygnalizację poprzez szlak JAK/STAT3. Bortezomib indukuje również odpowiedzi cytoprotekcyjne, takie jak zwiększenie ilości białek szoku cieplnego (np. hsp90), tak więc inhibitory tych białek cytoprotekcyjnych mogą zwiększać wrażliwość na bortezomib lub pokonywać oporność na lek.20 Pomimo omówionych wcześniej danych, swoiste efekty inhibicji proteasomów w nowotworach złośliwych i precyzyjny mechanizm działania bortezomibu pozostają niejasne i powinny być przedmiotem dalszych badań.
Bortezomib: farmakokinetyka i farmakodynamika
Farmakokinetyka i szlaki eliminacji bortezomibu nie zostały w pełni scharakteryzowane. Po podaniu dożylnym ponad 90% leku szybko znika z osocza w ciągu minut.1 Bortezomib jest metabilizowany przez mikrosomalne enzymy wątrobowe związane z cytochromem p450 na kilka nieczynnych metabolitów.41
Z powodu szybkiego usuwania leku z krwi, stworzono test biologiczny w celu oceny stopnia inhibicji proteasomów, pomocny w badaniach klinicznych fazy I i II. W badaniach na naczelnych wyznaczono, że docelowy poziom inhibicji proteasomów nie powinien przekraczać 80%. Przy zalecanym dawkowaniu, osiąga się około 60% inhibicji proteasomów. Stopień inhibicji proteasomów jest zależny od dawki.1
Monitorowanie inhibicji proteasomów nie jest konieczne w rutynowej praktyce klinicznej. Nie istnieją dobre dane odnośnie interakcji lekowych i farmakokinetyki u dzieci.
Bortezomib: Efekty uboczne i dawki
Najczęstszymi efektami toksycznymi przypisywanymi terapii bortezomibem są działania uboczne żołądkowo-jelitowe, przejściowa trombocytopenia, zmęczenie, gorączka i neuropatia obwodowa.3 Nudności, wymioty i wzdęcia mogą być objawami neuropatii wegetatywnej, lecz nie zostało to jeszcze dobrze zbadane. Większość z tych efektów toksycznych ma 1–2 stopień ciężkości. Mniej powszechne efekty uboczne obejmują wysypkę (15%), ból głowy (20%) i zawroty głowy (10%).
Zazwyczaj nie występują efekty uboczne związane z dożylnym wlewem leku, a rutynowa premedykacja nie jest konieczna. Zaleca się profilaktycznie środki przeciwwymiotne jeżeli pacjent doświadcza nudności lub wymiotów w trakcie terapii oraz podawanie roztworu izotonicznego chlorku sodu w celu zapewnienia właściwego nawodnienia. Gorączka występuje u około 20% pacjentów i jest zazwyczaj małego stopnia, ale czasami może osiągać 39?C lub więcej. Występuje często przy pierwszym cyklu terapii, około 12 godzin po podaniu leku, i trwa 24–26 godzin. Cytopenie (głównie leukopenia i trombocytopenia) są powszechne i leczy się zgodnie ze stardardami. U około 30% pacjentów może występować trombocytopenia 3 stopnia lub wyższego i może ona wymagać zmniejszenia dawki leku lub przetoczenia preparatu płytek krwi. W większości przypadków, jest ona przejściowa i przewidywalna (występuje zazwyczaj po 10 dniu). U niektórych pacjentów, proporcja zmniejszenia liczby płytek może być stała, prowadząc do mniejszych spadków bezwzględnych wraz ze zmniejszaniem się liczby płytek w trakcie terapii.
Neuropatia obwodowa występuje u około 35% pacjentów i jest częstsza u pacjentów, którzy otrzymywali uprzednią terapię neurotoksyczną i u chorych z istniejącą już wcześniej neuropatią.3,42 Neuropatia jest głównie czuciowa i może być 3 stopnia u około 10% pacjentów. Objawy neuropatii mogą być zmiejszone przez dostosowanie dawki i są zazwyczaj odwracalne po zaprzestaniu stosowania bortezomibu. Niedociśnienie ortostatyczne (prawdopodobnie zależne od dawki) występuje u około 10% pacjentów i wiąże się z odwodnieniem, współistniejącym leczeniem przeciwnadciśnieniowym lub dysfunkcją wegetatywną. Podawanie dożylne roztworu chlorku sodu w momencie podawania bortezomibu może być pomocne w odwodnieniu. Następstwa niedociśnienia ortostatycznego mogą być poważne u chorych z istniejącymi już wcześniej stanami małej pojemności minutowej serca. Nie istnieją żadne dane farmakokinetyczne odnośnie pacjentów z upośledzeniem czynności nerek lub wątroby.41 Pacjentów z istotnym upośledzeniem czynności nerek (klirens kreatyniny 10–30 ml/min) włączano do badań nad MM i nie wydaje się by niewydolność nerek miała wpływ na skuteczność, toksyczność lub stopień inhibicji proteasomów.88 Bortezomib jest jednakże metabolizowany przez wątrobowe enzymy związane z cytochoromem p450 i zaleca się ostrożność przy stosowaniu leku u chorych z chorobą wątroby. Zwykła dawka bortezomibu w leczeniu nawrotów MM wynosi 1,3 mg/2, podawana 2 razy w tygodniu (w dni 1, 4, 8 i 11) co 21 dni.3 U pacjentów z działaniami niepożądanymi przy standardowej dawce bortezomibu można zmniejszyć dawkę do 1 mg/m2 i 0,7 mg/m2.41
Połączenie bortezomibu z innymi lekami chemioterapeutycznymi
W badaniach przedklinicznych oporność nowotworu na konwencjonalne leki chemioterapeutyczne może być pokonana poprzez dołączenie bortezomibu, co podkreśla znaczenie rozwoju badań nad takimi połączeniami w MM i innych nowotworach złośliwych.19 W dwóch opisanych wcześniej badaniach fazy II w MM, dodawano deksametazon u 106 pacjentów, u których nie udało się uzyskać odpowiedzi na leczenie lub którzy mieli chorobę postępującą przy terapii lekiem pojedynczym – bortezomibem.3,44 Dziewiętnastu spośród tych pacjentów (18%) odpowiedziało pozytywnie na dołączenie deksametazonu. W tej grupie znalazło się trochę pacjentów, którzy byli uprzednio oporni na kortykosteroidy, co sugeruje działanie, co najmniej addytywne.
We wstępnych doniesieniach dotyczących połączenia bortezomibu z melfalanem w MM relacjonowano obiecującą aktywność, ale konieczne jest zmniejszenie dawek obu leków.45 Wstępne wyniki sugerują, że połączenie bortezomibu z pegylowaną doksorubicyną również zasługuje na dodatkowe zbadanie.46
W nowotworach litych, istnieją silne dowody przedkliniczne na to, że aktywność bortezomibu jest istotnie wyższa, gdy stosuje się go w połączeniu z lekami chemioterapeutycznymi, takimi jak gemcytabina, doksorubicyna, irynotekan, docetaksel i paklitaksel.47 Wstępne wyniki z badań fazy I wykazują, że połączenie bortezomibu z innymi lekami chemioterapeutycznymi jest wykonalne i bezpieczne dla chorych.47 Aktualnie trwają badania kliniczne fazy II nad bortezomibem w połączeniu z gemcytabiną, docetakselem, irynotekanem i innymi lekami cytotoksycznymi.
Przyszłe kierunki
Bortezomib jest pierwszym inhibitorem proteasomów, który wszedł do praktyki klinicznej. Jak również jest pierwszym nowym lekiem przeciw-MM zatwierdzonym przez FDA w ciągu kilku ostatnich lat. Ukończono właśnie zbieranie materiału w międzynarodowym, wieloośrodkowym badaniu z randomizacją, porównującym bortezomib z pulsami deksametazonu w nawrotach MM lub MM lekoopornym. Trwające badania oceniają rolę w różnych nowotworach złośliwych samego bortezomibu i w połączeniach z innymi substancjami czynnymi.
Sukces stwierdzany w przypadku bortezomibu jest nadzwyczajny, ponieważ udowodniono na jego przykładzie, że proteasomy są nowym i uzasadnionym celem w leczeniu nowotworów. Jesteśmy pełni nadziei, że inne, bardziej ulepszone inhibitory tego układu enzymatycznego zostaną niedługo zbadane w badaniach klinicznych.
Tabela 1. Wybrane białka, na które wpływa inhbicja szlaku ubikwityna-proteasomy
Table 1. Selected proteins affected by inhibition of the ubiquitin-proteasome pathway
| Białka | Efekty inhibicji proteasomów |
| I-?B | Zwiększenie stężeń I-?B jest przyczyną inhibicji aktywności NF-?B, co prowadzi do inhibicji wzrostu, apoptozy i zmniejszenia ekspresji cytokin angiogennych i cząsteczek adhezyjnych |
| p21, p27 (inhibitory kinazy zależnej od cyklin); p15, p16, p18, p19 (rodzina Ink inhibitorów kinazy 4/6 zależnej od cykliny-D) | Zwiększenie stężeń tych inhibitorów kinazy zależnej od cyklin prowadzi do zatrzymania cyklu komórki w fazie G1-S i apoptozy |
| p53 | Zwiększenie supresora nowotworowego p53 prowadzi do apoptozy na drodze kilku mechanizmów, w tym zwiększenia aktywności p21 i Bax |
| Bax | Zwiększenie ilości Bax prowadzi do apoptozy, przezwyciężając efekty nadmiernej ekspresji Bcl-2 |
| c-myc, N-myc | Nie jest jasne, w jaki sposób nadmierna ekspresja produktów tych onkogenów sprzyja działaniu przeciwnowotworowemu; jedną z hipotez jest to, że występowanie sygnałów sprzecznych, w związku z równoczesnymi sygnałami apoptotycznymi pochodzącymi od innych białek, prowadzi ostatecznie do apoptozy |
| Cykliny | Sprzeczne sygnały dla komórki w wyniku zwiększenia stężeń cyklin A, B, D i E w sposób nieuporządkowany, oraz w połączeniu z równoczesnym zwiększeniem stężeń inhibitorów kinaz zależnych od cyklin, promują apoptozę |
| Uszkodzone białka komórkowe | Akumulacja tych białek, prawidłowo usuwanych przez proteasomy, może wywoływać apoptozę |
| JNK | Aktywacja JNK prowadzi do aktywacji kaspazy-8 i kaspazy-3; może również uwalniać cytochrom C |
| Topoizomeraza II? | Stężenie tego enzymu uwalniającego DNA od naprężeń związanych ze skręceniem jest regulowane podczas cyklu komórkowego; inhibicja proteasomów wpływa na tę regulację |
Skróty: I-?B (inhibitor czynnika jądrowego-kappa B), NF-?B (czynnik jądrowy-kappa B), JNK (N-końcowa kinaza c-Jun)
Tabela 2. Badania fazy II z bortezomibem
Table 2. Phase II trials with bortezomib
| Typ nowotworu | Liczba ocenianych pacjentów | Schemat dawkowania | Odsetek odpowiedzi (%) | Piśmiennictwo |
| NawrĂłt szpiczaka mnogiego/szpiczak mnogi lekooporny | 193 | 1,3 mg/m2, dni 1, 4, 8 i 11 co 21 dni | 35 | Richardson i wsp.3 |
| NawrĂłt szpiczaka mnogiego | 54 | 1,0 mg/m2 (28 pacjentĂłw) vs 1,3 mg/m2 (26 pacjentĂłw), dni 1, 4, 8 i 11 co 21 dni | 33 (1,0 mg/m2) 50 (1,3 mg/m2) | Berenson i wsp.24 |
| Nawrót chłoniaka/chłoniak lekooporny | 11 | 1,5 mg/m2, dni 1, 4, 8 i 11 co 21 dni | 45* | Goy i wsp.27 |
| Nawrót/oporny na leczenie o przebiegu powolnym/chłoniak z komórek strefy płaszcza | 14 | 1,5 mg/m2, dni 1, 4, 8 i 11 co 21 dni | 50 | O’Connor i wsp.28 |
| Rak nerki | 18 | 1,5–1,7 mg/m2 dwa razy w tygodniu przez 2 tygodnie, co 21 dni | 6 | Davis i wsp.29 |
| Rak nerki | 24 | 1,3–1,5 mg/m2 dwa razy w tygodniu przez 2 tygodnie, co 21 dni | 13 | Drucker i wsp.30 |
| Nawracający/przerzutowy mięsak tkanek miękkich | 11 | 1,5 mg/m2 dwa razy w tygodniu przez 2 tygodnie, co 21 dni | Zbyt wcześnie na ocenę; jak na razie brak pacjentów odpowiadających | Maki i wsp.31 |
| Zaawansowany rak niedrobnokomĂłrkowy oskrzela | 8 | 1,5 mg/m2, dni 1, 4, 8 i 11 co 21 dni | 12,5 | Stevenson i wsp.32 |
*Wszystkich pięciu pacjentów, którzy odpowiedzieli miało chłoniak z komórek strefy płaszcza
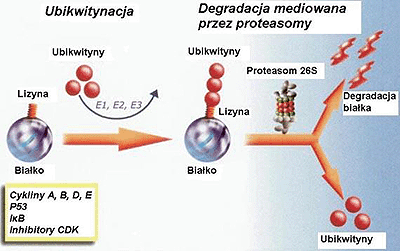
Rycina 1. Szlak ubikwityna-proteasomy. Na początku następuje dołączenie łańcuchów poliubikwitynowych do swoistych lizynowych grup czynnościowych białka przeznaczonego do zniszczenia (ubikwitynacja). Angażuje to trzy enzymy: enzym aktywujący ubikwitynę (E1), enzym koniugujący ubikwitynę (E2) i ligazę ubikwityny (E3). Ubikwitynowane białka są degradowane przez wewnątrzkomórkowe proteasomy 26S. I-?B, inhibitor czynnika jądrowego-kappa B; CDK, kinaza zależna od cyklin.
Figure 1. The ubiquitin-proteasome pathway. First is the addition of polyubiquitinated tails to specific lysine moieties on the protein destined for destruction (ubiquitination). This involves three enzymes, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). Ubiquitinated proteins are degraded by the intracellular 26S proteasome. I-B, nuclear factor-kappa B inhibitor; CDK, cyclin-dependant kinase.
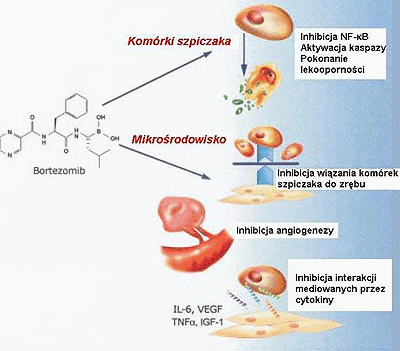
Rycina 2. Mechanizm działania bortezomibu w szpicaku mnogim. Inhibicja szlaku proteasomów przez bortezomib wywiera efekty zarówno na komórki szpiczaka, jak i na ich mikrośrodowisko. NF-?B, czynnik jądrowy-kappa B; IL-6, interleukina-6; VEGF, naczyniowy śródbłonkowy czynnik wzrostu; TNF-?, czynnik martwicy nowotworów alfa; IGF-1, insulino-podobny czynnik wzrostu-1.
Figure 2. Mechanism of action of bortezomib in multiple myeloma. Inhibition of the proteasome pathway by bortezomib has effects on both the myeloma cell and its microenvironment. NF-B, nuclear factor-kappa B; IL-6, interleukin-6; VEGF, vascular endothelial growth factor; TNF, tumor necrosis factor alpha; IGF-1, insulin-like growth factor-1.
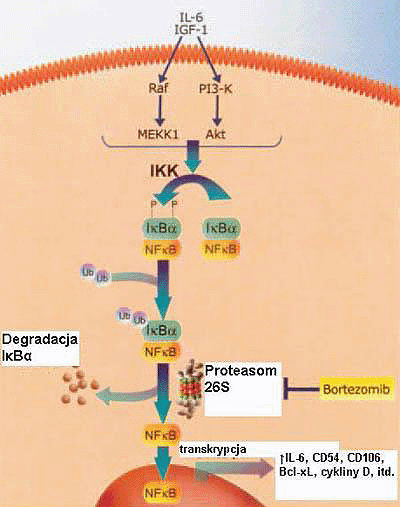
Rycina 3. Bortezomib i inhibicja czynnika jÄ…drowego-kappa B (NF-?B). Stymulacja komĂłrek nowotworowych przez czynniki wzrostu prowadzi do aktywacji kinazy (IKK) inhibitora (I-?B) NF-?B, ktĂłra fosforyluje (P) I-?B. Ufosforylowany I-?B jest ubikwitynowany (Ub) i degradowany w szlaku proteasomĂłw, uwalniajÄ…c NF-?B. Bortezomib zapobiega degradacji I-?B i tym samym inhibuje aktywacjÄ™ NF-?B. IL-6, interleukina-6; IGF-1, insulino-podobny czynnik wzrostu-1.
Fig 3. Bortezomib and nuclear factor-kappa B (NF-B) inhibition. Stimulation of tumor cells by growth factors leads to activation of NF-B inhibitor (I-B) kinase (IKK), which phosphorylates (P) I-B. Phosphorylated I-B is ubiquitinated (Ub) and degraded by the proteasome pathway releasing NF-B. Bortezomib prevents the degradation of I-B and, thereby, inhibits NF-B activation. IL-6, interleukin-6; IGF-1, insulin-like growth factor-1
Publikacja wraz z piśmiennictwem dostępna w gabinecie dra Artura Jurczyszyna. Zainteresowanych proszę o kontakt osobisty.
Artur Jurczyszyn, Klinika Hematologii, Szpital Uniwersytecki, KrakĂłw
Aleksander B. Skotnicki, Kierownik Katedry i Kliniki Hematologii CM UJ w Krakowie
Katedra i Klinika Hematologii Collegium Medicum Uniwersytetu Jagiellońskiego