
INHIBITORY PROTEASOM√ďW W LECZENIU SZPICZAKA MNOGIEGO
Streszczenie
Pomimo nieustannego postńôpu w terapii i diagnostyce szpiczaka mnogiego, choroba ta pozostaje wcińÖŇľ nieuleczalna. WcińÖŇľ pilnie potrzebne sńÖ nowe sposoby leczenia ukierunkowane, zar√≥wno na kom√≥rki nowotworowe (patologiczne plazmocyty), jak i na mikroŇõrodowisko szpiku kostnego. Przedkliniczne badania in vitro oraz in vivo wykazujńÖ niezwykŇāńÖ aktywnoŇõńá przeciw-nowotworowńÖ podczas zastosowania inhibitora proteasom√≥w ‚Äď bortezomibu (PS-341); nawet w przypadku kom√≥rek szpiczaka mnogiego opornych na uprzednio stosowane terapie, w tym deksametazon, melfalan i talidomid. W oparciu o te wyniki UrzńÖd ds. ŇĽywnoŇõci i Rejestracji Lek√≥w USA (FDA) zatwierdziŇā niedawno pierwszy inhibitor proteasom√≥w, znany uprzednio jako PS-341 ‚Äď bortezomib (Velcade) do leczenia nawrotowego i opornego szpiczaka mnogiego. Badania genomiki i proteomiki dostarczajńÖ dalszych racjonalnych podstaw dla poŇāńÖczenia bortezomibu z konwencjonalnymi i nowymi lekami, w celu zahamowania wzrostu szpiczaka mnogiego, pokonania opornoŇõci na leki, zmniejszenia towarzyszńÖcych terapii dziaŇāaŇĄ niepoŇľńÖdanych i poprawienia wynik√≥w leczenia.
Wprowadzenie
Kom√≥rki szpiczaka mnogiego umiejscawiajńÖ sińô gŇā√≥wnie w szpiku kostnym, gdzie r√≥Ňľnorodne czynniki humoralne wspomagajńÖ wzrost i przeŇľycie kom√≥rek nowotworowych, a takŇľe zapobiegajńÖ efektom cytotoksycznym chemioterapii.1 Przyleganie kom√≥rek szpiczaka mnogiego do kom√≥rek zrńôbu szpiku kostnego wywoŇāuje wydzielanie cytokin, takich jak interleukina-6 (IL-6)2 oraz insulinopodobny czynnik wzrostu (IGF-1) lub naczyniowo Ňõr√≥dbŇāonkowy czynnik wzrostu (VEGF), kt√≥re z kolei nie tylko indukujńÖ proliferacjńô kom√≥rek szpiczaka mnogiego, ale takŇľe hamujńÖ wywoŇāanńÖ przez chemioterapińô apoptozńô kom√≥rek nowotworowych.1,3,4 MikroŇõrodowisko szpiku kostnego ma istotny udziaŇā w patogenezie i progresji szpiczaka mnogiego, a nowe leki przeciw-nowotworowe, ukierunkowane zar√≥wno na patologiczne plazmocyty, jak i ich mikroŇõrodowisko, majńÖ ogromne zastosowanie kliniczne.
PomyŇõlny rozw√≥j leczenia bortezomibem (PS-341) w szpiczaku mnogim pokazaŇā, Ňľe hamowanie proteasom√≥w jest skutecznńÖ strategińÖ terapeutycznńÖ.5 Analog dipeptydu kwasu boronowego ‚Äď bortezomib jest silnym, wysoce wybi√≥rczym i odwracalnym inhibitorem proteasom√≥w, ukierunkowanym na kompleks proteasomowy 26S i hamujńÖcym jego czynnoŇõńá (Rycina 1). Proteasom 26S jest zaleŇľnńÖ od ATP, posiadajńÖcńÖ wiele miejsc katalitycznych proteazńÖ mediujńÖcńÖ degradacjńÖ biaŇāek wewnńÖtrzkom√≥rkowych. Degradacja proteasomowa biaŇāek Ňļle sfaŇādowanych lub uszkodzonych nastńôpuje poprzez podjednostkńô regulatorowńÖ 16S proteazy 26S, wielokrotnie ubikwitynowanych biaŇāek i ich nastńôpowńÖ hydrolizńô na maŇāe polipeptydy (Rycina 1). Poza eliminowaniem biaŇāek uszkodzonych i Ňļle sfaŇādowanych, proteasomy regulujńÖ takŇľe kluczowe procesy kom√≥rkowe, w tym modulacjńô czynnik√≥w transkrypcyjnych, postńôp cyklu kom√≥rkowego, zatrzymanie wzrostu i apoptozńô. Nasz artykuŇā zwraca szczeg√≥lnńÖ uwagńô na: (a) przedkliniczne i kliniczne dane o hamowaniu proteasom√≥w jako sposobie leczenia w szpiczaku mnogim; (b) aktywnoŇõńá cytotoksycznńÖ poŇāńÖczenia bortezomibu z innymi konwencjonalnymi lub nowymi lekami przeciw szpiczakowi mnogiemu; i (c) strategie pokonywania opornoŇõci na bortezomib w kom√≥rkach nowotworowych, w tym oparte na genomice i proteomice molekularne sposoby leczenia, jak r√≥wnieŇľ na ocenńô nowych inhibitor√≥w proteasom√≥w.
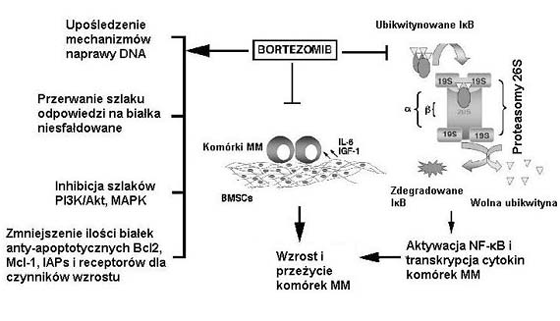
Rycina 1. Bortezomib/PS-341 wpŇāywa na r√≥Ňľne szlaki wzrostu i przeŇľycia w kom√≥rkach szpiczaka mnogiego (MM). Leczenie kom√≥rek szpiczaka mnogiego bortezomibem wińÖŇľe sińô z nastńôpujńÖcymi zdarzeniami: inhibicjńÖ przylegania kom√≥rek szpiczaka mnogiego do kom√≥rek zrńôbowych szpiku kostnego (BMSCs), co prowadzi zahamowania zaleŇľnej od przylegania transkrypcji i wydzielania licznych cytokin; inhibicjńÖ NF-?B; upoŇõledzeniem mechanizm√≥w naprawy DNA; zmniejszeniem aktywnoŇõci sygnalizacyjnych szlak√≥w wzrostowych i anty-apoptotycznych i iloŇõci zwińÖzanych z nimi biaŇāek, takich jak kinazy biaŇākowej aktywowanej przez mitogeny (MAPK), kinazy 3-fosfatydyloiznozytolu (PI3K)/Akt, Bcl2 czy inhibitor√≥w biaŇāek apoptozy (IAPs).
Budowa i regulacja proteasomów
Proteasomy sńÖ kluczowymi regulatorami degradacji biaŇāek.6 Kompleks proteasomowy 26S posiada 2 jednostki 19S i poŇāoŇľony pomińôdzy nimi, baryŇākowaty rdzeŇĄ.7 Cztery uŇāoŇľone razem pierŇõcienie skŇāadajńÖ sińô na strukturńô 20S: dwa centralne pierŇõcienie ? otoczone sńÖ przez 2 inne pierŇõcienie, z kt√≥rych kaŇľdy skŇāada sińô z siedmiu biaŇāek (Rycina 1). Proteasomowa degradacja biaŇāek nastńôpuje na drodze nastńôpujńÖcych po sobie zdarzeŇĄ: biaŇāko zostaje oznakowane ŇāaŇĄcuchem maŇāego polipeptydu lub ubikwitynńÖ; nastńôpnie enzym estryfikujńÖcy ubikwitynńô E1 aktywuje i wińÖŇľe jńÖ z enzymem E2 sprzńôgajńÖcym ubikwitynńô w spos√≥b zaleŇľny od ATP; ligaza E3 ubikwityny przyczepia czńÖsteczkńô ubikwityny do biaŇāka; tworzy sińô dŇāugi ŇāaŇĄcuch polipeptydowy zŇāoŇľony z czńÖsteczek ubikwitiny i ostatecznie nastńôpuje degradacja przez proteasomy biaŇāka na maŇāe fragmenty.7,8
Degradacja biaŇāek mediuje zar√≥wno prawidŇāowńÖ czynnoŇõńá kom√≥rki, jak r√≥wnieŇľ odpowiedŇļ kom√≥rkowńÖ na chemioterapińô.9 W licznych badaniach wykazano, Ňľe ubikwitynacja biaŇāek i degradacja poprzez szlaki ubikwityna-proteasomy reguluje postńôp cyklu kom√≥rkowego, supresjńô nowotworu, transkrypcjńô, replikacjńô DNA, zapalenie i apoptozńô.10 Mutacje lub zmiany w tych szlakach sygnaŇāowych prowadzńÖ do upoŇõledzenia przechodzenia z fazy G1 do S.11 Inhibitory proteasom√≥w hamujńÖ degradacjńô biaŇāek i powodujńÖ akumulacjńô biaŇāek Ňļle sfaŇādowanych lub uszkodzonych, co z kolei wywoŇāuje odpowiedŇļ szoku cieplnego i Ňõmierńá kom√≥rki.7,12 BiorńÖc pod uwagńô, Ňľe szlak ubikwityna-proteasomy wpŇāywa na wiele proces√≥w kom√≥rkowych, jego zahamowanie z zastosowaniem inhibitora proteasom√≥w wpŇāywa na szersze spektrum biaŇāek o r√≥Ňľnorodnych funkcjach.
Proteasomy i leczenie przeciwnowotworowe
Liczne badania wykazujńÖ, iŇľ inhibitory proteasom√≥w sńÖ bardziej cytotoksyczne dla proliferujńÖcych kom√≥rek nowotworu zŇāoŇõliwego niŇľ spoczynkowych kom√≥rek prawidŇāowych.13 Prawdopodobnie, kom√≥rki nowotworu zŇāoŇõliwego majńÖ zmienione lub upoŇõledzone biaŇāka cyklu kom√≥rkowego, co prowadzi do zwińôkszenia szybkoŇõci proliferacji, zwińôkszenia gromadzenia sińô uszkodzonych biaŇāek i w ten spos√≥b do wińôkszej zaleŇľnoŇõci od proces√≥w degradacji proteosomalnej. Co waŇľne, bortezomib wywoŇāuje apoptozńô w licznych kom√≥rkach szpiczaka mnogiego w dawkach, kt√≥re nie wpŇāywajńÖ na ŇľywotnoŇõńá prawidŇāowych limfocyt√≥w.5 Ponadto, czynnik jńÖdrowy-?B (nuclear factor-?B ‚Äď NF-?B) wińÖŇľe sińô z proliferacjńÖ i opornoŇõcińÖ na leki w kom√≥rkach nowotworowych (w tym szpiczaka mnogiego),14 a bortezomib zmniejsza aktywacjńÖ NF-?B, nasilajńÖc tym samym cytotoksyczne efekty chemioterapii (Rycina 1).5,12,15 Wyniki te sugerujńÖ, Ňľe proteasomy sńÖ waŇľnym celem dla chemioterapii, przy tolerowalnym wskaŇļniku terapeutycznym.
Indukowana przez bortezomib apoptoza koreluje z osŇāabionńÖ aktywnoŇõcińÖ NF-?B
Konstytutywna aktywacja NF-?B wińÖŇľe sińô ze wzrostem/proliferacjńÖ i opornoŇõcińÖ na leki, nadajńÖc w ten spos√≥b kom√≥rkom nowotworowym i prawidŇāowym r√≥ŇľnicujńÖcńÖ wraŇľliwoŇõńá na inhibitory proteasom√≥w.15 Aktywacja NF-?B nastńôpuje na drodze sekwencji nastńôpujńÖcych zdarzeŇĄ: wywoŇāywana przez nadrzńôdnńÖ kinazńô I?B? fosforylacja I?B; ubikwitynacja i degradacja ufosforylowanego I?B? prowadzńÖca do powstania wolnego kompleksu p50/60 i przemieszczenie do jńÖdra i aktywacja p50/60 NF-?B.16 Gdy tylko znajdzie sińô w jńÖdrze, NF-?B wińÖŇľe sińô ze zgodnymi sekwencjami obecnymi w regionach promotorowych wielu gen√≥w zwińÖzanych z czynnikami wzrostu/przeŇľycia i wywoŇāuje ich transkrypcjńô. Na przykŇāad, aktywacja NF-?B sprzyja wytwarzaniu cytokin (IL-6, TNF-?), czynnik√≥w przeŇľycia (inhibitory biaŇāek apoptozy i Bcl-XI), i czńÖsteczek przylegania mińôdzykom√≥rkowego (czńÖsteczka przylegania mińôdzykom√≥rkowego, czńÖsteczka przylegania kom√≥rek naczyniowych i selektyna-E).16 Wszystkie te czńÖsteczki uŇāatwiajńÖ wzrost i przeŇľycie kom√≥rek nowotworowych. NF-?B poŇõredniczy w kluczowych czynnoŇõciach kom√≥rki, w tym w odpowiedzi immunologicznej, jak r√≥wnieŇľ podczas wzrostu, przeŇľycia i apoptozy kom√≥rek szpiczakowych.17 Wewnńôtrzna aktywacja NF-?B wińÖŇľe sińô z wzrostem/przeŇľyciem licznych kom√≥rek szpiczaka mnogiego. Przyleganie patologicznych plazmocyt√≥w do kom√≥rek zrńôbowych szpiku kostnego wywoŇāuje mediowanńÖ przez NF-?B transkrypcjńô i wydzielanie IL-6 i IGF-1.17,18 Zar√≥wno IL-6, jak i IGF-1 sprzyjajńÖ przeŇľyciu kom√≥rek szpiczaka mnogiego w szpiku kostnym, poprzez hamowanie apoptozy wywoŇāywanej przez leki konwencjonalne, takie jak deksametazon. Ponadto, kom√≥rki nowotworowe pacjenta majńÖ zwińôkszonńÖ (w por√≥wnaniu do kom√≥rek prawidŇāowych) aktywnoŇõńá NF-?B.19 Odwrotnie, wraŇľliwe na leki kom√≥rki szpiczaka mnogiego wykazujńÖ mniejszńÖ aktywnoŇõńá NF-?B niŇľ oporne na leczenie kom√≥rki szpiczaka mnogiego, co sugeruje, Ňľe NF-?B nadaje opornoŇõńá na chemioterapińô.19 Zwińôkszone stńôŇľenia NF-?B relacjonowano takŇľe w kom√≥rkach szpiczaka mnogiego pochodzńÖcych od chorych z nawrotem po chemioterapii.17 ŇĀńÖcznie, wyniki te wskazujńÖ, Ňľe NF-?B jest kluczowym regulatorem wzrostu i przeŇľycia kom√≥rek nowotworowych w Ňõrodowisku szpiku kostnego. Co waŇľne, leczenie szpiczaka mnogiego z zastosowaniem bortezomibu zapobiega degradacji I?B, hamujńÖc tym samym nie tylko aktywacjńô NF-?B, ale takŇľe zwińÖzane z tym wytwarzanie cytokin (Rycina 1). JednakŇľe jest maŇāo prawdopodobne, Ňľe samo hamowanie NF-?B odpowiada za caŇāoŇõciowńÖ aktywnoŇõńá bortezomibu przeciw szpiczakowi mnogiemu.20 Na przykŇāad, zar√≥wno bortezomib, jak i swoisty inhibitor I?B ‚Äď PS-1145 hamujńÖ aktywacjńô NF-?B, jednakŇľe w przeciwieŇĄstwie do bortezomibu, PS-1145 jedynie czńôŇõciowo hamuje wzrost kom√≥rek szpiczaka mnogiego (zahamowanie 20‚Äď40% przez PS-1145 w por√≥wnaniu do 80‚Äď90% przez bortezomib),20 co sugeruje, Ňľe bortezomib posiada w kom√≥rkach szpiczaka mnogiego dodatkowe cele, nie tylko NF-?B.
Bortezomib aktywuje plejotropowe szlaki sygnaŇāowe
W badaniach biochemicznych in vitro wykazano obecnie, Ňľe apoptoza indukowana przez bortezomib wińÖŇľe sińô z nastńôpujńÖcymi dodatkowymi zdarzeniami (Rycina 2): (a) pobudzenie klasycznych biaŇāek odpowiedzi stresowej, takich jak biaŇāka szoku cieplnego Hsp27, Hsp70 i Hsp90;21 (b) zwińôkszenie iloŇõci NH2-koŇĄcowej kinazy c-jun;22 (c) zmiana potencjaŇāu bŇāony mitochondrialnej i wytwarzanie reaktywnych odmian tlenu;23 (d) indukcja wewnńôtrznego szlaku Ňõmierci kom√≥rki (tj. uwolnienie do cytozolu biaŇāek mitochondrialnych ‚Äď cytochromu c i drugiego mitochndrialnego aktywatora kaspaz i aktywacja kaskady kaspaza-9 > kaspaza-3); (e) aktywacja zewnńôtrznej sygnalizacji apoptotycznej przez cińôcie Bid i kaspazy-8; (f) upoŇõledzenie mechanizm√≥w naprawy DNA na drodze inaktywacji zaleŇľnej od DNA kinazy biaŇākowej;24 (g) zahamowanie przylegania licznych kom√≥rek szpiczaka mnogiego do kom√≥rek zrńôbowych szpiku kostnego i wińÖŇľńÖcego sińô z tym wydzielania cytokin;25 i (h) hamowanie szlak√≥w sygnaŇāowych kinazy biaŇākowej aktywowanej przez mitogeny i kinazy 3-fosfatydyloiznozytolu/Akt.26 Wszystkie powyŇľsze mechanizmy sygnaŇāowe mogńÖ mieńá ŇāńÖczny udziaŇā w dziaŇāaniu bortezomibu przeciw szpiczakowi mnogiemu. W szczeg√≥lnoŇõci stwierdzono obligatoryjnńÖ rolńô aktywacji NH2-koŇĄcowej kinazy c-jun podczas indukowanej bortezomibem apoptozy szpiczaka mnogiego, potwierdzonńÖ zastosowaniem strategii mutacji dominujńÖco-ujemnych lub swoistych biochemicznych inhibitor√≥w NH2-koŇĄcowej kinazy c-jun.22 Wynik ten zostaŇā niedawno potwierdzony przez inne badanie w kom√≥rkach niedrobnokom√≥rkowego raka oskrzela.27
Poza wymienionymi powyŇľej wydarzeniami sygnaŇāowymi, hamowanie proteasom√≥w wpŇāywa takŇľe na biaŇāka regulujńÖce cykl kom√≥rkowy, takie jak gen supresorowy (nowotwor√≥w) TP 53 (p53). Zmiany w p53 prowadzńÖ do niestabilnoŇõci genetycznej w wielu r√≥Ňľnorodnych kom√≥rkach nowotworowych.28 W kontekŇõcie szpiczaka mnogiego, wykazano, Ňľe bortezomib wywoŇāuje apoptozńô w dzikich (pod wzglńôdem p 53), jak i zmutowanych kom√≥rkach nowotworowych.12 Wyniki te sńÖ sp√≥jne z innymi badaniami na kom√≥rkach raka okrńôŇľnicy, glejaka i biaŇāaczkowych.29 Co wińôcej, apoptoza indukowana przez bortezomib w kom√≥rkach szpiczaka mnogiego koreluje z fosforylacjńÖ p53 (Ser15).26 Inne badanie wykazaŇāo, Ňľe leczenie kom√≥rek raka gruczoŇāu krokowego LNCaP-Pro5 bortezomibem wińÖŇľe sińô z: (a) stabilizacjńÖ p53 bez fosforylacji Ser15 i Ser20, a p53 pozostaje zwińÖzane ze swoim inhibitorem MDM2; (b) translokacjńÖ p53 do jńÖdra i zwińôkszonym wińÖzaniem sińô p53 do DNA, akumulacjńÖ transkrypt√≥w zaleŇľnych od p53, jak r√≥wnieŇľ aktywacjńÖ gen√≥w reporterowych odpowiedzi na p53; i (c) zmniejszeniem indukowanej przez bortezomib aktywacji p53 i Ňõmierci kom√≥rek.30 Nie okreŇõlono, czy mutacje p53 wpŇāywajńÖ na indukowanńÖ przez bortezomib cytotoksycznoŇõńá.
Prawdopodobnie, na cytotoksycznoŇõńá indukowanńÖ przez bortezomib, wpŇāywajńÖ mutacje w COOH-koŇĄcowej domenie p53, zawierajńÖcej gŇā√≥wne miejsce dziaŇāania dla ligazy ubikwityny. Badania grupy Kennetha Andersona i wsp. sugerujńÖ, Ňľe bortezomib zabija kom√≥rki bez wzglńôdu na stan ich zmutowania. Do zbadania pozostaje jednakŇľe, czy miejsca mutacji p53 w kom√≥rkach szpiczaka mnogiego w rzeczywistoŇõci stanowińÖ gŇā√≥wne miejsca ligacji ubikwityny, czy nie. Bardziej szczeg√≥Ňāowe badanie z zastosowaniem konstrukt√≥w ze zmutowanym p53 (zwŇāaszcza zawierajńÖcych mutacje w domenie COOH-koŇĄcowej) dostarczńÖ danych zwińÖzanych z istotnoŇõcińÖ p53 podczas indukowanej przez bortezomib apoptozy w kom√≥rkach szpiczaka mnogiego. Og√≥lnie rzecz biorńÖc, wyniki w r√≥Ňľnych typach nowotwor√≥w sugerujńÖ, Ňľe apoptoza indukowana przez bortezomib nastńôpuje zar√≥wno w spos√≥b zaleŇľny, jak i niezaleŇľny od p53.
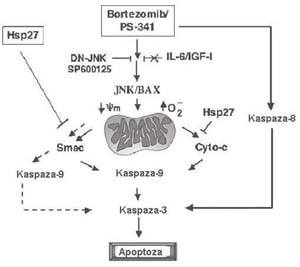
Rycina 2. Sygnalizacja apoptotyczna wywoŇāana przez bortezomib/PS-341. Bortezomib indukuje aktywacjńô NH2-koŇĄcowej kinazy c-jun (JNK), kt√≥ra przemieszcza sińô do mitochondri√≥w i uŇāatwia uwalnianie cytochromu c (Cyto-c) i drugiego mitochondrialnego aktywatora kaspaz (Smac) z mitochondri√≥w do cytozolu, z nastńôpowńÖ aktywacjńÖ kaspazy-9. Bortezomib aktywuje takŇľe kaspazńô-8. Zar√≥wno kaspaza-8, jak i kaspaza-9 indukujńÖ aktywacjńô swojego podrzńôdnego efektora ‚Äď kaspazy-3 i cińôcie polimerazy poli(ADP-rybozy) (PARP). Blokada NH2-koŇĄcowej kinazy c-jun z zastosowaniem dominujńÖco-ujemnej NH2-koŇĄcowej kinazy c-jun (DN-JNK) lub inhibitora biochemicznego SP600125 znosi hamuje uwalnianie cytochromu c/ Smac i aktywacjńô kaspazy-9. Apoptoza indukowana przez bortezomib nie jest hamowana przez IL-6 lub insulino-podobny czynnik wzrostu-1 (IGF-1). Ektopowa ekspresja Hsp27 hamuje wywoŇāane przez bortezomib uwalnianie cytochromu c i Smac
DziaŇāanie przeciwnowotworowe bortezomibu in vivo
StosujńÖc mysi model przeszczepu ksenogenicznego ludzkiego szpiczaka, zbadano skutecznoŇõńá, toksycznoŇõńá i mechanizm dziaŇāania bortezomibu in vivo.31 Zaobserwowano wyraŇļne zahamowanie wzrostu nowotworu u myszy leczonych bortezomibem. Mediana caŇākowitego przeŇľycia byŇāa r√≥wnieŇľ przedŇāuŇľona w spos√≥b istotny w por√≥wnaniu z grupńÖ kontrolńÖ. Bortezomib byŇā dobrze tolerowany w dawce 0,5 mg/kg (i.v.), ale niekt√≥re myszy leczone dawkńÖ 1,0 mg/kg staŇāy sińô umierajńÖce i traciŇāy masńô ciaŇāa. Analiza nowotwor√≥w uzyskanych leczonych zwierzńÖt wykazaŇāa, Ňľe bortezomib indukowaŇā apoptozńô i zmniejszaŇā angiogenezńô. Og√≥lnie rzecz biorńÖc, wyniki te wykazujńÖ, Ňľe bortezomib wywiera in vivo istotne dziaŇāanie przeciw szpiczakowi mnogiemu w dawkach dobrze tolerowanych w modelu mysim, co potwierdza badanie in vitro. W innym badaniu z zastosowaniem przeszczep√≥w ksenogenicznych LOVO wykazano,32 Ňľe terapia ŇāńÖczona bortezomibem i CPT-11 prowadziŇāa do wyraŇļnego zwińôkszenia poziom√≥w apoptozy i regresji nowotworu w por√≥wnaniu do leczenia kaŇľdym z lek√≥w osobno, co sugerujńÖc istotny potencjaŇā bortezomibu w poŇāńÖczeniu z innymi chemioterapeutykami w celu nasilaniu aktywnoŇõci przeciwnowotworowej, zmniejszaniu toksycznoŇõci i pokonywaniu opornoŇõci na leki.
Badania kliniczne nad bortezomibem
Badania przedkliniczne in vitro wykazujńÖce aktywnoŇõńá bortezomibu przeciw kom√≥rkom szpiczaka mnogiego zostaŇāy potwierdzone w badaniach fazy I, w nowotworach hematologicznych i litych.33,34 Podczas wstńôpnego badania okreŇõlajńÖce zakres dawek u chorych z opornym na leczenie szpiczakiem mnogim, chŇāoniakiem i biaŇāaczkńÖ, pacjenci otrzymywali bortezomib we wstrzyknińôciach i.v. 2 razy w tygodniu przez 4 tygodnie z nastńôpowymi 2 tygodniami bez leczenia. Maksymalna dawka tolerowana wynosiŇāa 1,04 mg/m2.33 Efektami toksycznymi ograniczajńÖcymi dawkńô byŇāy zmńôczenie i zŇāe samopoczucie, trombocytopenia, i zaburzenia r√≥wnowagi elektrolitowej. Badania fazy I wykazaŇāy zachńôcajńÖce odpowiedzi u chorych ze szpiczakiem mnogim: jednńÖ odpowiedŇļ caŇākowitńÖ (CR), stwierdzonńÖ na podstawie ujemnego wyniki immunofiksacji; oraz osiem odpowiedzi ze zmniejszeniem biaŇāka monoklonalnego w surowicy i naciek√≥w nowotworowych w szpiku kostnym. Co wińôcej, przeciwnowotworowńÖ aktywnoŇõńá bortezomibu w tych badaniach fazy I stwierdzono r√≥wnieŇľ w przypadku chŇāoniaka nieziarnieczego (NHL).
Inne badanie fazy I oceniaŇāo skutecznoŇõńá bortezomibu w zaawansowanych nowotworach litych, przy zastosowaniu 3-tygodniowego cyklu dawkowania (dwa razy w tygodniu przez 2 tygodnie, z nastńôpowym 1 tygodniem bez leczenia).34 Maksymalna dawka tolerowana wynosiŇāa 1,56 mg/m2, co sugeruje, Ňľe cykl 3-tygodniowy moŇľe pozwalańá na podawanie wińôkszych dawek niŇľ cykl 6-tygodniowy. Nie obserwowano Ňľadnych hematologicznych efekt√≥w toksycznych ograniczajńÖcych dawkńô, a niehematologiczne efekty toksyczne ograniczajńÖce dawkńô obejmowaŇāy neuropatińô 3-ego stopnia i biegunkńô. Ponadto, neuropatińô 3-ego stopnia obserwowano gŇā√≥wnie u pacjent√≥w z uprzednimi dowodami neuropatii, a po przerwaniu podawania leku neuropatia ulegaŇāa poprawie. W koŇĄcu, bortezomib wykazywaŇā aktywnoŇõńá przeciwnowotworowa takŇľe w innych nowotworach zŇāoŇõliwych, takich jak niedrobnokom√≥rkowy rak oskrzela, raki noso-gardŇāa, czerniak zŇāoŇõliwy i rak nerki.34
Badania fazy II w szpiczaku mnogim
Badania fazy II obejmowaŇāy pacjent√≥w z nawrotem szpiczaka mnogiego/szpiczakiem mnogim opornym na leczenie.35 KaŇľdy cykl leczenia obejmowaŇā bortezomib (1,3 mg/m2) podawany 2 razy w tygodniu, z 1 tygodnie bez leczenia. Podawano 8 cykli leczenia u os√≥b odpowiadajńÖcych na terapińô, a pacjenci z odpowiedziami suboptymalnymi otrzymywali doustnie deksametazon po pierwszych 2 cyklach bortezomibu. WŇāńÖczono pacjent√≥w (n = 202), z kt√≥rych wszyscy otrzymywali kortykosteroidy, 92% leki alkilujńÖce, 81% antracykliny, 83% talidomid, i 64% przeszczep kom√≥rek pnia. Mediana liczby uprzednich terapii wynosiŇāa szeŇõńá. SpoŇõr√≥d 193 chorych, 4% osińÖgnńôŇāo CR, stwierdzonńÖ na podstawie braku wykrycia biaŇāka M, zar√≥wno w elektroforezie, jak i immunofiksacjńÖ; 6% osińÖgnńôŇāo prawie-CR, stwierdzonńÖ w oparciu o zastosowaniu immunofiksacji; 18% i 7% wykazywaŇāo odpowiedni odpowiedzi czńôŇõciowe (PR) i minimalne (MR), przy caŇākowitym odsetku odpowiedzi 35% (CR+PR+MR). Mediana przeŇľycia dla caŇāej populacji wynosiŇāa 16 miesińôcy, a chorzy osińÖgajńÖcy duŇľńÖ odpowiedŇļ (CR+PR) przeŇľywali istotnie dŇāuŇľej niŇľ ci, kt√≥rzy jej nie osińÖgali. SpoŇõr√≥d 74 pacjent√≥w, kt√≥rzy nie osińÖgnńôli co najmniej MR i w zwińÖzku z tym otrzymywali deksametazon w poŇāńÖczeniu z bortezomibem, u 18% stwierdzono poprawńô ‚Äď obejmowaŇāo to 6 pacjent√≥w z chorobńÖ opornńÖ na deksametazon, dostarczajńÖc dowod√≥w na to, Ňľe bortezomib moŇľe pokonywańá opornoŇõńá na deksametazon. Powszechnymi, efektami ubocznymi leczenia byŇāy nudnoŇõci, wymioty, biegunka, zmńôczenie, utrata apetytu (w tym jadŇāowstrńôt), zaparcia, neuropatia obwodowa, gorńÖczka, niedokrwistoŇõńá i trombocytopenia.
W innym otwartym badaniu fazy II nad bortezomibem,36 54 pacjent√≥w ze szpiczakiem mnogim, kt√≥rzy mieli nawr√≥t po lub byli oporni na leczenie pierwszego rzutu poddano randomizacji do grup otrzymujńÖcych i.v. 1,0 lub 1,3 mg/m2 bortezomibu dwa razy w tygodniu przez 2 tygodnie, co 3 tygodnie, przez maksymalnie osiem cykli. Deksametazon byŇā dozwolony u pacjent√≥w u chorych z chorobńÖ postńôpujńÖcńÖ/stabilnńÖ po odpowiednio 2 lub 4 cyklach. Odsetek CR+PR dla samego bortezomibu wynosiŇā 30% i 38%, odpowiednio w grupach otrzymujńÖcych 1,0 mg/m2 (8 spoŇõr√≥d 27 pacjent√≥w) i 1,3 mg/m2 (10 spoŇõr√≥d 26 pacjent√≥w). Odsetek CR+PR u chorych otrzymujńÖcych sam bortezomib lub w poŇāńÖczeniu z deksametazonem wynosiŇā w kohortach leczonych dawkami 1,0 i 1,3 mg/m2 odpowiednio 37% i 50%. Najpowszechniejszymi dziaŇāaniami niepoŇľńÖdanymi 3-go stopnia byŇāy trombocytopenia (24%), neutropenia (17%), limfopenia (11%) i neuropatia obwodowa (9%). DziaŇāania 4-go stopnia obserwowano u 9% pacjent√≥w (5 spoŇõr√≥d 54). Sam bortezomib lub w poŇāńÖczeniu z deksametazonem wykazywaŇā aktywnoŇõńá przeciw szpiczakowi mnogiemu u chorych z nawrotami po terapii pierwszego rzutu.
Niedawno, w pierwszym i najwińôkszym badaniu z randomizacjńÖ (APEX), przeprowadzonym w 93 OŇõrodkach w Ameryce P√≥Ňānocnej, Europie i Izraelu, wykazano wińôkszńÖ skutecznoŇõńá bortezomibu jako leku pojedynczego w por√≥wnaniu z wysokimi dawkami deksametazonu u chorych z nawrotem szpiczaka mnogiego.37 Przy stosowaniu bortezomibu obserwowano istotny okres czasu wolnego od progresji i korzyŇõńá w przeŇľyciu w por√≥wnaniu do deksametazonu. Przewagńô obserwowano takŇľe, zar√≥wno u chorych otrzymujńÖcych terapińô drugiego rzutu, jak i p√≥Ňļne leczenie ratujńÖce. Profile bezpieczeŇĄstwa bortezomibu i deksametazonu byŇāy moŇľliwe do przewidzenia, wzglńôdnie zr√≥wnowaŇľone z moŇľliwymi do opanowania efektami toksycznymi.
TrwajńÖce badania nad bortezomibem w poŇāńÖczeniu jak r√≥wnieŇľ jako lekiem pojedynczym wykazujńÖ obiecujńÖcńÖ aktywnoŇõńá i korzystny profil dziaŇāaŇĄ ubocznych. Co ciekawe, adriamycyna+deksametazon+bortezomib wykazywaŇāy duŇľy odsetek odpowiedzi, ale przy wińôkszej toksycznoŇõci. Odwrotnie, bortezomib jako lek pojedynczy wykazywaŇā minimalnńÖ toksycznoŇõńá z mniejszym odsetkiem odpowiedzi.37 Wstńôpne wyniki z badania grupy francuskiej wykazujńÖ skutecznoŇõńá poŇāńÖczenia bortezomib + deksametazon jako schematu indukcyjnego przed przeszczepem autologicznym kom√≥rek pnia u chorych z nowo rozpoznanym szpiczakiem mnogim, przy maŇāej toksycznoŇõci.38 Wyniki te, ŇāńÖcznie z obserwowanymi przez Jagganath‚Äôa i wsp., potwierdzajńÖ iŇľ poŇāńÖczenie bortezomib/deksametazon jest skuteczne i dobrze tolerowane u chorych z nowo rozpoznanym szpiczakiem mnogim. Podczas XI International Myeloma Workshop, kt√≥re odbyŇāy sińô w Sydney doniesiono o bardzo obiecujńÖcych wstńôpnych pr√≥bach ŇāńÖczenia Bortezomibu oraz Relvimidu, osińÖgajńÖc odpowiedŇļ pozytywnńÖ u ponad 90% pacjent√≥w z MM.
Rozw√≥j opornoŇõci na bortezomib i strategie terapeutyczne pokonywania opornoŇõci na bortezomib
Bortezomib zabija kom√≥rki szpiczaka mnogiego, jednakŇľe przedŇāuŇľona ekspozycja wińÖŇľe sińô z toksycznoŇõcińÖ i rozwojem opornoŇõci na bortezomib. Dla pokonania opornoŇõci na lek, podstawowe znaczenie ma zbadanie jej mechanizmu. Grupa Kennetha Andersona wykazaŇāa, Ňľe opornoŇõńá na chemioterapińô w kom√≥rkach szpiczaka mnogiego pojawia sińô na skutek nastńôpujńÖcych wydarzeŇĄ: (a) nadmierna ekspresja glikoproteiny P; (b) biaŇāka przeciwapoptotyczne, takie jak Bcl2 lub inhibitory biaŇāek apoptozy; (c) defekty apoptotycznych szlak√≥w sygnaŇāowych indukowanych przez leki, takich jak te wystńôpujńÖce na poziomie mitochondri√≥w lub siateczki Ňõr√≥dplazmatycznej; (d) zwińôkszona ekspresja receptor√≥w czynnik√≥w wzrostu i zwińÖzanych z nimi szlak√≥w sygnaŇāowych; i, na koniec, (e) interakcja pomińôdzy kom√≥rkami szpiczaka mnogiego i mikroŇõrodowiskiem szpiku kostnego gospodarza. W rzeczy samej, jest maŇāo prawdopodobne by jeden swoisty mechanizm wywoŇāywaŇā odpowiedŇļ na bortezomib, a prawdopodobne, Ňľe suma wielu r√≥Ňľnorodnych czynnik√≥w moŇľe prowadzińá do rozwoju opornoŇõci na lek.
Badania profilu genetycznego i badania proteomiczne z zastosowaniem bortezomibu oraz innych lek√≥w przeciw szpiczakowi mnogiemu dostarczyŇāy podstawy dla ŇāńÖczenia lek√≥w w celu zabicia opornych na leczenie kom√≥rek szpiczaka mnogiego. Na przykŇāad, nasze badania in vitro wykazaŇāy, Ňľe poŇāńÖczenie bortezomibu z innymi lekami konwencjonalnymi, takimi jak deksametazon, doksorubicyna, melfalan lub mitoksantron, wywoŇāuje addytywnńÖ i(lub) synergistycznńÖ aktywnoŇõńá przeciwszpiczakowńÖ.5,19,24 Co wińôcej, terapia ŇāńÖczona kom√≥rek szpiczaka mnogiego kom√≥rek pacjent√≥w ze szpiczakiem mnogim bortezomibem i nowymi lekami, takimi jak relvimid lub triterpenoidem ‚Äď CDDO-imidazolidem, wywoŇāuje synergistycznńÖ aktywnoŇõńá przeciw szpiczakowi mnogiemu nawet w pochodzńÖcych od pacjent√≥w kom√≥rkach szpiczaka mnogiego pacjent√≥w opornych na bortezomib,50,66 dostarczajńÖc tym samym podstawy dla protokoŇā√≥w klinicznych stosujńÖcych ten schemat leczenia.39 Te ŇāńÖczone strategie zmniejszńÖ towarzyszńÖcńÖ toksycznoŇõńá i pokonajńÖ i(lub) zapobiegnńÖ rozwojowi opornoŇõci lekowej. W wielooŇõrodkowym badaniu SUMMIT, 35% wstńôpnie silnie leczonych pacjent√≥w z nawrotem szpiczaka mnogiego/szpiczakiem mnogim opornym na leczenie odpowiedziaŇāo na monoterapińô bortezomibem, a efekty toksyczne byŇāy moŇľliwe do opanowania. PoŇāńÖczenie deksametazonu z bortezomibem wywoŇāywaŇāo dodatkowe odpowiedzi u chorych z suboptymalnymi odpowiedziami na bortezomib, co potwierdza podobne, addytywne, hamujńÖce efekty tych lek√≥w na kom√≥rki szpiczaka mnogiego w badaniach in vitro. Ponadto, w oparciu o dane przedkliniczne, kilka trwajńÖcych obecnie badaŇĄ ocenia przeciwnowotworowńÖ aktywnoŇõńá bortezomibu w poŇāńÖczeniu z melfalanem, pegylowanńÖ doksorubicynńÖ liposomalnńÖ (Doxil) i talidomidem.40 Jak do dzisiaj, dane wykazujńÖ silnńÖ aktywnoŇõńá przeciw-nowotworowńÖ bortezomibu w poŇāńÖczeniu z innymi lekami u chorych ze szpiczakiem mnogim, przy moŇľliwych do opanowania efektach toksycznych.
Niedawne badania dostarczajńÖ takŇľe danych o biaŇākach, kt√≥re nadajńÖ kom√≥rkom szpiczaka mnogiego opornoŇõńá na bortezomib. Na przykŇāad, badanie wykazaŇāo, Ňľe leczenie bortezomibem indukuje apoptozńô w kom√≥rkach chŇāoniaka SUDHL6 (DHL6), ale nie SUDHL4 (DHL4).21 Analiza mikromacierzy wykazaŇāa duŇľe stńôŇľenia RNA dla biaŇāka szoku cieplnego 27 (Hsp27) w kom√≥rkach DHL4 w por√≥wnaniu do DHL6. Inhibicja Hsp27 z zastosowaniem strategii antysensownej przywraca wraŇľliwoŇõńá na bortezomib w kom√≥rkach DHL4. Odwrotnie, nadmierna ekspresja Hsp27 typu dzikiego sprawia, Ňľe wraŇľliwe na bortezomib kom√≥rki DHL6 staja sińô na niego oporne. Dane te dostarczajńÖ dowod√≥w, Ňľe Hsp27 nadaje opornoŇõńá na bortezomib. DuŇľe stńôŇľenia Hsp27 stwierdzono takŇľe w kom√≥rkach szpiczaka mnogiego pozyskanych od pacjent√≥w opornych na leczenie bortezomibem. Konieczne sńÖ dalsze badania dla ustalenia, czy hamowanie Hsp27 z zastosowaniem specyficznych dla stopnia klinicznego inhibitor√≥w nasila dziaŇāanie bortezomibu przeciw szpiczakowi mnogiemu i pokonuje opornoŇõńá lekowńÖ. Pomimo to, w oparciu o te wyniki, wydaje sińô, iŇľ naleŇľy ukierunkowańá dziaŇāanie na p38MAPK ‚Äď nadrzńôdny aktywator Hsp27 w celu zahamowania wzrostu kom√≥rek szpiczaka mnogiego. Wyniki wykazujńÖ, Ňľe inhibicja p38MAPK nasila aktywnoŇõńá bortezomibu przeciw szpiczakowi mnogiemu.41 Wypracowano juŇľ protok√≥Ňā kliniczny wykorzystujńÖcy inhibitor p38MAPK razem z bortezomibem u pacjent√≥w ze szpiczakiem mnogim. Wiadomo, Ňľe bortezomib wywiera swoje dziaŇāania poprzez hamowanie proteasom√≥w kom√≥rkowych, jednakŇľe niejasne pozostaje to, czy inhibicja proteasom√≥w jest zawsze wymagana dla apoptozy indukowanej przez bortezomib. Wyniki grupy Kennetha Andersona wykazaŇāy, Ňľe leczenie bortezomibem prowadziŇāo do odpowiednio 82% i 88% inhibicji aktywnoŇõci proteasom√≥w w kom√≥rkach chŇāoniaka, zar√≥wno opornych na bortezomib SUDHL4, jak i wraŇľliwych na bortezomib SUDHL6.21 ŇĀńÖcznie dane te potwierdzajńÖ, Ňľe: (a) szlak inhibicji proteasom√≥w nie jest wadliwy w opornych na bortezomib kom√≥rkach DHL4 i (b) inhibicja proteasom√≥w nie koreluje z apoptozńÖ. BezpoŇõrednie okreŇõlenie inhibicji proteasom√≥w w pr√≥bkach krwi i tkanek pacjenta badano w badaniach fazy I. Bortezomib byŇā dobrze tolerowany w dawkach prowadzńÖcych inhibicji proteasom√≥w do 80%.42 Ponadto, przedŇāuŇľone podawanie nie powodowaŇāo dalszego zmniejszenia wraŇľliwoŇõci na inhibicjńô proteasom√≥w. ŇĀńÖcznie, dane te sugerujńÖ, Ňľe inhibicja proteasom√≥w jest gŇā√≥wnńÖ czynnoŇõcińÖ inhibitora proteasom√≥w, ale Ňľe hamowanie proteasom√≥w moŇľe nie korelowańá ze stopniem cytotoksycznoŇõci w kom√≥rkach nowotworowych.
Opr√≥cz Hsp27, r√≥wnieŇľ czŇāonkowie rodziny biaŇāka Bcl2 przyczyniajńÖ sińô do rozwoju opornoŇõci lekowej w wielu typach kom√≥rek,43 a apoptoza wywoŇāywana przez bortezomib w kom√≥rkach szpiczaka mnogiego jest r√≥wnieŇľ czńôŇõciowo znoszona przez ekspresjńô Bcl2.44 Zwińôkszona ekspresja inhibitor√≥w biaŇāek apoptozy, takich jak XIAP, r√≥wnieŇľ moŇľe przyczyniańá sińô do opornoŇõci na bortezomib.44 TrwajńÖce badania przedkliniczne badajńÖ r√≥Ňľne leki lub swoiste inhibitory biochemiczne, kt√≥re hamujńÖ czynnoŇõńá tych biaŇāek, wywoŇāujńÖc tym samym apoptozńô nawet w opornych na leki kom√≥rkach szpiczaka mnogiego.
Wnioski
Inhibicja proteasom√≥w okazaŇāa sińô silnńÖ strategińÖ terapeutycznńÖ w leczeniu nawrot√≥w szpiczaka mnogiego lub choroby opornej na leczenie. Bortezomib jest pierwszym od ponad dekady lekiem zatwierdzonym przez FDA do leczenia szpiczaka mnogiego. R√≥Ňľnorodne badania kliniczne oceniajńÖ aktualnie bortezomib w innych typach nowotwor√≥w. Ponadto, badania kliniczne bortezomibu w poŇāńÖczeniu z innymi lekami chemioterapeutycznymi pomagajńÖ stworzyńá nowe strategie terapeutyczne w szpiczaku mnogim. KoŇĄczńÖc, przedkliniczna ocena innego, nowego inhibitora proteasom√≥w wykazuje istotnńÖ aktywnoŇõńá przeciw szpiczakowi mnogiemu, nawet przeciw kom√≥rkom szpiczaka opornego na bortezomib, przy niŇľszych towarzyszńÖcych efektach ubocznych dla kom√≥rek prawidŇāowych. Dostarcza to podstaw dla protokoŇā√≥w klinicznych do pokonania opornoŇõci na bortezomib i poprawienia wynik√≥w leczenia chorego.
Publikacja wraz z piŇõmiennictwem dostńôpna w gabinecie dra Artura Jurczyszyna. Zainteresowanych proszńô o kontakt osobisty.
Artur Jurczyszyn, Klinika Hematologii, Szpital Uniwersytecki, Kraków
Aleksander B. Skotnicki, Kierownik Katedry i Kliniki Hematologii CM UJ w Krakowie
Katedra i Klinika Hematologii Collegium Medicum Uniwersytetu JagielloŇĄskiego