MECHANIZMY PATOGENICZNE WARUNKUJńĄCE NOWE SPOSOBY TERAPII W SZPICZAKU MNOGIM – ZNACZENIE CYTOKIN ‚Äď czńôŇõńá 1
‚ÄěPathogenetic mechanisms conditioning novel therapeutic ways for multiple myeloma ‚Äď significance of cytokines‚ÄĚ ‚Äď part 1
Streszczenie:
Szpiczak mnogi (MM) jest zŇāoŇõliwńÖ, do tej pory wcińÖŇľ nieuleczalnńÖ chorobńÖ nowotworowńÖ, kt√≥rej istotńÖ jest powolny rozrost monoklonalnych plazmocyt√≥w lub plazmoblast√≥w w szpiku kostnym. W Stanach Zjednoczonych na te chorobńô zapada rocznie okoŇāo 15 000 nowych pacjent√≥w. CzńôstoŇõńá wystńôpowania MM w USA wynosi okoŇāo 3 na 100.000 mieszkaŇĄc√≥w, przewaŇľajńÖ mńôŇľczyŇļni (60%), zaŇõ 98% chorych ma > 40 lat. Wstńôpne badania in vitro na zwierzńôtach sugerujńÖ rolńô interakcji kom√≥rek nowotworowych z podŇõcieliskiem szpiku kostnego w regulacji ich wzrostu, migracji i opornoŇõci na leki antyproliferacyjne. Strategie terapeutyczne, ukierunkowane na mechanizmy, na drodze, kt√≥rych kom√≥rki MM wzrastajńÖ i proliferujńÖ w szpiku kostnym, w tym talidomid oraz jego silne pochodne immunomodulujńÖce, jak r√≥wnieŇľ inhibitor proteasom√≥w PS-341, sńÖ w stanie przezwycińôŇľańá opornoŇõńá na leki w wielu prowadzonych badaniach klinicznych.
SŇāowa kluczowe: szpiczak mnogi, cytokiny, talidomid, PS-341
Abstract:
Multiple myeloma (MM) is a disseminated neoplasm of terminally differentiated plasma cells that is incurable with currently available therapies. MM affects about 15.000 new patients annually in the USA. Preliminary in vitro and animal studies suggest a role for MM-host interactions in regulating MM cell growth, drug resistance and migration in the bone marrow. Importantly, treatment strategies which target mechanisms whereby MM cells grow and proliferation in the bone marrow, including thalidomide and its potent immunomodulatory derivatives and proteasome inhibitor PS-341, can overcome classical drug resistance in clinical studies.
Key words: multiple myeloma, cytokines, thalidomide, PS-341
Aktualne sposoby leczenia szpiczaka mnogiego
Konwencjonalne sposoby terapii obejmujńÖce Ňõrodki alkilujńÖce, antracykliny i kortykosteroidy sńÖ w stanie przedŇāuŇľyńá okres przeŇľycia chorego na szpiczaka mnogiego (MM) Ňõrednio do 3-4 lat. (1) Zastosowanie terapii wysokodozowanej z nastńôpowym autologicznym przeszczepieniem kom√≥rek macierzystych szpiku kostnego moŇľe wydŇāuŇľyńá ŇõrednińÖ przeŇľycia do 4-5 lat. (2) Ulepszania procedury autoprzeszczep√≥w obejmujńÖ zar√≥wno powtarzalne zastosowanie terapii wysokodozowanych, (3) jak r√≥wnieŇľ immunologiczne strategie leczenia minimalnej choroby resztkowej po wykonaniu przeszczepienia. (4) ChociaŇľ wyniki terapii ulegŇāy duŇľej poprawie w kilku ostatnich latach, jednak wyleczyńá udaje sińô niewielu, jeŇõli w og√≥le jakichŇõ pacjent√≥w. Terapia wysokodozowana i alloprzeszczep szpiku kostnego wińÖŇľńÖ sińô wcińÖŇľ z duŇľńÖ ŇõmiertelnoŇõcińÖ zwińÖzanńÖ z procedurńÖ przeszczepowńÖ. (5) Obecnie trwajńÖ wysiŇāki obejmujńÖce wykorzystanie tego sposobu podejŇõcia na wczeŇõniejszym etapie przebiegu choroby, (6) usunińôcie limfocyt√≥w T z alloprzeszczep√≥w w celu uniknińôcia reakcji przeszczep przeciwko gospodarzowi (7) i wykorzystanie terapii nie mieloablacyjnej w celu zmniejszenia toksycznoŇõci (tzw. minialloprzeszczep) (8) Postńôpy w leczeniu podtrzymujńÖcym obejmujńÖ zar√≥wno wykorzystanie bisfosfonian√≥w, (9) znaczńÖco zmniejszajńÖcych powikŇāania kostne i poprawiajńÖcych jakoŇõńá Ňľycia, jak i stworzenie rekombinowanych czynnik√≥w wzrostu wpŇāywajńÖcych pobudzajńÖco na hematopoezńô. Pomimo wielu postńôp√≥w, MM pozostaje w dalszym cińÖgu chorobńÖ nieuleczalnńÖ m.in. z powodu rozwoju opornoŇõci kom√≥rek nowotworowych na wszystkie konwencjonalne sposoby leczenia, co zwraca uwagńô na naglńÖcńÖ potrzebńô nowych strategii terapeutycznych.
Rola mikroŇõrodowiska szpiku kostnego w patogenezie choroby
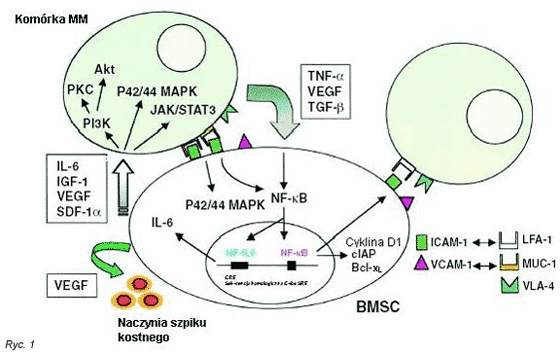
MM jest B-kom√≥rkowym zŇāoŇõliwym nowotworem charakteryzujńÖcym sińô patologicznym, niekontrolowanym rozrostem monoklonalnych kom√≥rek plazmatycznych w szpiku kostnym, w powińÖzaniu z obecnoŇõcińÖ biaŇāka monoklonalnego w surowicy i/lub moczu, zmniejszonymi stńôŇľeniami prawidŇāowych immunoglobulin (Ig) oraz chorobńÖ litycznńÖ koŇõci. Liczne dowody molekularne sugerujńÖ, Ňľe kom√≥rki MM sńÖ transformowanymi odpowiednikami plozmoblast√≥w/plazmocyt√≥w centr√≥w postgerminalnych w szpiku kostnym. (10) MikroŇõrodowisko szpiku kostnego odgrywa istotnńÖ rolńô w nasilaniu wzrostu, przeŇľywalnoŇõci, migracji i opornoŇõci na leki kom√≥rek szpiczakowych11 oraz okreŇõlaniu mechanizm√≥w poŇõredniczńÖcych w patogenezie nowotworu (Ryc. 1). Kom√≥rki MM zagnieŇľdŇľajńÖ sińô i przylegajńÖ do biaŇāek macierzy poza-kom√≥rkowej oraz do kom√≥rek zrńôbowych podŇõcieliska szpiku kostnego, co nie tylko umiejscawia je w nowym Ňõrodowisku, ale niesie r√≥wnieŇľ waŇľne nastńôpstwa czynnoŇõciowe. (12) WińÖzanie kom√≥rek MM z fibronektyna- biaŇākiem macierzy poza-kom√≥rkowej wywoŇāuje prawdopodobnie opornoŇõńá na leki, co jest zwińÖzane z przyleganiem kom√≥rek ze sobńÖ (CAM-DR) (13) oraz zwińôkszeniem biaŇāka p27. PowyŇľsze zjawiska zachodzńÖ niezaleŇľnie od cytokin. Ponadto, liczne badania scharakteryzowaŇāy zar√≥wno wińÖzanie sińô kom√≥rek MM z podŇõcieliskiem szpiku kostnego, jak i tego nastńôpstwa, w tym wzrost i opornoŇõńá na leki. (12) Przyleganie kom√≥rek MM do podŇõcieliska szpiku nie tylko w spos√≥b zbliŇľony poŇõredniczy w opornoŇõci na wywoŇāywanńÖ przez leki apoptozńô, ale takŇľe wywoŇāuje wydzielanie w spos√≥b parakrynny interleukiny-6 (IL-6), najwaŇľniejszej cytokiny poŇõredniczńÖcej we wzroŇõcie i przeŇľyciu kom√≥rek MM. (14)
Wykazano r√≥wnieŇľ, Ňľe umiejscowione w Ňõrodowisku szpiku kostnego kom√≥rki nowotworowe wydzielajńÖ cytokiny [czynnik martwicy nowotwor√≥w-őĪ (TNF-őĪ), transformujńÖcy czynnik wzrostu-ő≤ (TGF-ő≤), czynnik wzrostu Ňõr√≥dbŇāonka naczyniowego (VEGF) insulinopodobny czynnik wzrostu-1 (IGF-1) i czynnik pochodzńÖcy z kom√≥rek zrńôbowych (SDF)-1őĪ], kt√≥re nastńôpnie zwińôkszajńÖ wytwarzanie IL-6 w kom√≥rkach zrńôbu szpiku kostnego. (15,16,17) (Ryc. 1). Kaskady sygnaŇāowe poŇõredniczńÖce w tych dziaŇāaniach cytokin mogńÖ stanowińá cele nowatorskich strategii terapeutycznych.
Rola cytokin w szpiczaku mnogim
Interleukina – 6 (IL-6) i rozpuszczalna forma receptora dla Interleukiny – 6 (sIL-6R)
IL-6 i rozpuszczalna forma receptora dla interleukiny-6 (sIL-6R) peŇānińÖ kluczowńÖ rolńô w proliferacji plazmocyt√≥w szpiczakowych (18-20). W warunkach prawidŇāowych IL-6 wyzwala r√≥Ňľnicowanie limfocyt√≥w B i jest najwaŇľniejszym czynnikiem podtrzymujńÖcym zdolnoŇõci ich przeŇľycia (21,22). IL-6 powoduje przeksztaŇācenie nieufosforylowanej postaci biaŇāka retinoblastoma (pRB) w aktywnńÖ postańá ufosforylowanńÖ, co prowadzi do uwolnienia czynnika transkrypcyjnego E2F1 i proliferacji kom√≥rek szpiczakowych. Rolńô genu RB w szpiczaku mnogim podkreŇõla sińôgajńÖca 70% czńôstoŇõńá zar√≥wno mutacji genu jak i obecnoŇõci jego produktu biaŇāka RB (23). W przeciwieŇĄstwie do oddziaŇāywania na prawidŇāowe plazmocyty IL-6 wzbudza proliferacjńô kom√≥rek szpiczakowych in vitro i in vivo, ale nie indukuje wydzielania immunoglobulin (24).
Zasadniczym mechanizmem dziaŇāania IL-6 jest najprawdopodobniej hamowanie apoptozy kom√≥rki szpiczakowej. IL-6 obficie wydzielana przez kom√≥rki nowotworowe zaangaŇľowana jest w patogenezńô niekt√≥rych zmian patologicznych typowych dla MM. Wykazuje bezpoŇõrednie dziaŇāanie osteoklastyczne oraz uszkadzajńÖce nerki, sprzyjajńÖc powstaniu tzw. ‚Äěnerki szpiczakowej‚ÄĚ (25). IL-6 moŇľe byńá produkowana w spos√≥b autokrynny przez nowotworowe plazmocyty lub parakrynny przez kom√≥rki podŇõcieliska szpiku kostnego wzbudzone w wyniku adhezji kom√≥rek szpiczakowych (Ryc. 4) (26). Dla proliferacji plazmocytarnej podstawowe znaczenie ma parakrynna droga sekrecji IL-6. Mniej istotny patogenetycznie autokrynny spos√≥b wydzielania IL-6 zachodzi po zwińÖzaniu CD40L do powierzchniowego CD40 na plazmocytach, co powoduje przejŇõcie pRB do aktywnej ufosforylowanej postaci (27).
Receptor IL-6 skŇāada sińô z dw√≥ch podjednostek: ŇāaŇĄcucha a (CD126) oraz ŇāaŇĄcucha b – glikoproteiny gp 130 (CD130), stanowińÖcej wŇāaŇõciwy przekaŇļnik sygnaŇā√≥w do wnńôtrza kom√≥rki (47). Interleukina-6, jak i inne cytokiny z rodziny IL-6, o podstawowym znaczeniu dla rozrostu nowotworowego, ŇāńÖczńÖ sińô z antygenem CD126 i aktywujńÖ biaŇāko peŇānińÖce rolńô przekaŇļnika sygnaŇā√≥w gp130. W kom√≥rce szpiczakowej istniejńÖ dwie drogi wewnńÖtrzkom√≥rkowego przekazywania sygnaŇā√≥w: szlak JAK ‚Äď STAT-3 i szlak Ras ‚Äď MAPK. W tym pierwszym istotnńÖ rolńô odgrywajńÖ nieodŇāńÖcznie zwińÖzane z receptorem IL-6 kinazy tyrozynowe Janusa JAK1, JAK2 i TYK2, kt√≥re fosforylujńÖ kom√≥rkowe substraty ‚Äď czynniki transkrypcyjne STAT. Szczeg√≥lnńÖ rolńô odgrywa STAT-3, kt√≥ry po fosforylacji przechodzi do jńÖdra kom√≥rkowego i dziaŇāa jako czynnik inicjujńÖcy transkrypcjńô genu kodujńÖcego IL-6 (28). Szlak ten moŇľe byńá odpowiedzialny za wywoŇāywanie opornoŇõci kom√≥rki szpiczakowej na apoptozńô. UwaŇľa sińô, Ňľe bardziej istotna dla proliferacji nowotworowej jest druga ŇõcieŇľka transmisji sygnaŇā√≥w zwińÖzana z onkogenem Ras i kinazńÖ biaŇākowńÖ aktywowanńÖ przez mitogeny (MAPK) (29). R√≥Ňľnice w udziale odpowiednich kaskad przekazywania sygnaŇā√≥w w kom√≥rce mogńÖ przekŇāadańá sińô mińôdzy innymi na r√≥Ňľnice odpowiedzi niekt√≥rych kom√≥rek szpiczakowych na IL-6 (30).
Rozpuszczalna forma receptora dla IL-6 (sIL-6R), o masie czńÖsteczkowej 55 KD, zostaŇāa wyizolowana z surowicy krwi i moczu chorych na MM (31). Co ciekawe sIL-6R tworzńÖc kompleks z IL-6 zachowuje zdolnoŇõńá do wińÖzania sińô z gp130 i aktywacji kom√≥rki docelowej (32). Odwrotnie zachowuje sińô wińôkszoŇõńá pozostaŇāych rozpuszczalnych receptor√≥w cytokinowych, kt√≥re po poŇāńÖczeniu ze swym rozpuszczalnym ligandem tracńÖ swojńÖ aktywnoŇõńá. sIL-6R poszerza wińôc spektrum kom√≥rek, kt√≥re aktywowańá moŇľe IL-6, o kom√≥rki posiadajńÖce gp130, a nie posiadajńÖce IL-6R33. Wykazano, Ňľe stńôŇľenie sIL-6R w surowicy krwi jest podwyŇľszone u chorych na szpiczaka mnogiego w por√≥wnaniu do zdrowych os√≥b i koreluje z cińôŇľkoŇõcińÖ choroby. Aktualnie ocena stńôŇľenia sIL-6R w surowicy krwi uznawana jest za jeden z czynnik√≥w rokowniczych w MM34.
Czynnik martwicy nowotwor√≥w-őĪ (TNF-őĪ)
ChociaŇľ TNF-őĪ wydzielany przez kom√≥rki MM nie indukuje ich znaczńÖcego wzrostu, przeŇľycia lub opornoŇõci na leki, to wińÖŇľe sińô bezpoŇõrednio z regionem odpowiedzi na TNF-őĪ promotora IL-6 i silniej stymuluje wydzielanie IL-6 w podŇõcielisku szpiku kostnego niŇľ VEGF lub TGF-ő≤. (15) TNF-őĪ wydzielany przez kom√≥rki MM indukuje r√≥wnieŇľ zaleŇľnńÖ od NF-őļB zwińôkszonńÖ ekspresjńô czńÖsteczek adhezyjnych w kom√≥rkach MM i podŇõcielisku szpiku (CD49d i CD54). (15) W ten spos√≥b wińÖzanie kom√≥rek nowotworowych, transkrypcja i wydzielanie IL-6 w podŇõcielisku ulega zwińôkszeniu (Ryc 1). (15) Nowe leki, ukierunkowane na TNF-őĪ, w tym talidomid i jego pochodne immunomodulujńÖce, (35) mogńÖ dziaŇāańá, co najmniej czńôŇõciowo, poprzez hamowanie nastńôpstw poŇõredniczonych przez IL-6.
Czynnik wzrostu Ňõr√≥dbŇāonka naczyniowego (VEGF)
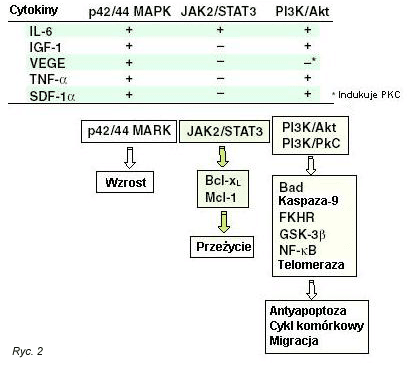
VEGF wytwarzany jest zar√≥wno przez kom√≥rki MM, jak i podŇõcielisku szpiku kostnego. MoŇľe odpowiadańá za zwińôkszenie angiogenezy obserwowane u chorych z MM. Aktywacja CD40 indukuje zaleŇľne od p53 wydzielanie VEGF w ludzkich kom√≥rkach szpiczakowych. (36) Niekt√≥re linie kom√≥rek MM i patologiczne plazmocyty pochodzńÖce od pacjent√≥w wykazujńÖ ekspresjńô receptora VEGF (VEGFR) Flt-1. Egzogenne VEGF wywoŇāuje fosforylacjńô Flt-1, (17) sŇāabe pobudzenie ERK, proliferacjńô kom√≥rek nowotworowych, co moŇľe byńá neutralizowane przez przeciwciaŇāa przeciwko VEGF, inhibitor kinazy tyrozynowej VEGFR PTK78737 lub przez PD98059. (17) GŇā√≥wnymi nastńôpstwami dziaŇāania VEGF jest migracja kom√≥rek MM, w wińôkszym stopniu u pacjent√≥w z biaŇāaczkńÖ plazmatycznokom√≥rkowńÖ (PCL), niŇľ w kom√≥rkach chorych z MM. Indukowana przez VEGF migracja kom√≥rek MM wińÖŇľe sińô z zaleŇľnńÖ od ő≤1-integryny i PI3K aktywacjńÖ PKCőĪ, ale nie z aktywacjńÖ ERK (Ryc. 2). BezpoŇõrednie dziaŇāania VEGF na kom√≥rki nowotworowe, indukowanie cytokin (IL-6) i angiogenezńô w Ňõrodowisku szpiku kostnego sugerujńÖ potencjalnńÖ uŇľytecznoŇõńá sposob√≥w leczenia skierowanych na VEGF (Ryc. 1), takich jak inhibitor kinazy tyrozynowej VEGFR PTK787. (37)
Czynnik wzrostu hepatocytów (HGF)
Czynnik wzrostu hepatocyt√≥w (HGF) ma wŇāaŇõciwoŇõci mitogenne, chemotaktyczne i morfogenne; dziaŇāa na kom√≥rki za poŇõrednictwem receptora powierzchniowego kodowanego przez prot-onkogen c-met. (38) HGF indukuje angiogenezńô poprzez stymulacjńô proliferacji, migracji i adhezji kom√≥rek Ňõr√≥dbŇāonka. Ponadto wywiera istotny wpŇāyw na morfogenezńô nowych naczyŇĄ krwionoŇõnych oraz pobudza podŇõcielisko szpiku kostnego do syntezy czynnik√≥w angiogennych. (38) PodwyŇľszone stńôŇľenia HGF w surowicy u chorych na MM zwińÖzane sńÖ z niekorzystnńÖ prognozńÖ. Liczne badania dowodzńÖ, iŇľ HGF zaangaŇľowany jest w proliferacji nowotworowej, uszkodzeniu koŇõci, angiogenezie oraz adhezji kom√≥rek szpiczakowych do podŇõcieliska w mikroŇõrodowisku szpiku kostnego. (39) Wydaje sińô, iŇľ bardzo istotnńÖ rolńô odgrywa mechanizm parakrynny i autokrynny stymulacji HGF i c-met. (40) Receptor c-met nie wystńôpuje na limfocytach B krwi obwodowej, natomiast jego obecnoŇõńá zostaŇāa potwierdzona w 100% kom√≥rek szpiczakowych na poziomie mRNA, a takŇľe na poziomie biaŇāka. (40) Najnowsze badania wskazujńÖ na istotnńÖ rolńô osi HGF-c-met w stymulacji proliferacji oraz zahamowaniu apotozy kom√≥rek MM. W badaniach wŇāasnych (41) analizowano 76 pacjent√≥w ze szpiczakiem mnogim leczonych w Klinice Hematologii Szpitala Uniwersyteckiego w Krakowie. BiorńÖc pod uwagńô chorych z progresjńÖ oraz bez progresji MM wyr√≥Ňľniono, iŇľ osoczowe stńôŇľenie HGF to optymalny marker prognostyczny r√≥ŇľnicujńÖcy pacjent√≥w. W grupie odpowiadajńÖcej na zastosowane leczenie (bez progresji choroby) wykazano osoczowe stńôŇľenia HGF o wartoŇõciach 1283 ¬Ī 1583 pg/ml, zaŇõ w grupie z progresjńÖ choroby (wykazujńÖcej opornoŇõńá na leczenie) uzyskano wartoŇõci 3555 ¬Ī 4498 pg/ml, r√≥Ňľnica ta byŇāa istotna statystycznie (p<0,001). W badanich wŇāasnych ocena osoczowego stńôŇľenia HGF metodńÖ krzywych ROC pozwoliŇāa na zdiagnozowanie braku odpowiedzi na zastosowane leczenie z czuŇāoŇõcińÖ 94,4 % i swoistoŇõcińÖ 73,9 % przy osoczowych wartoŇõciach stńôŇľeŇĄ powyŇľej 1048 pg/ml. W zwińÖzku z powyŇľszym wydaje sińô, iŇľ stńôŇľenie HGF w osoczu krwi to dobry marker r√≥ŇľnicujńÖcy chorych ze szpiczakiem mnogim, jeŇõli chodzi o zastosowane leczenie antyproliferacyjne i rokowanie na przyszŇāoŇõńá. (41)
Insulinopodobny czynnik wzrostu-1 (IGF-1)
IGF-1 sprzyja proliferacji i opornoŇõci na leki w kom√≥rkach MM poprzez aktywacjńô kaskad sygnaŇāowych MAPK i PI3K/Akt. (42) IGF-1 stymuluje utrzymujńÖce sińô pobudzenie NF-őļB i PI3/Akt, indukuje fosforylacjńô biaŇāka FKHR, reguluje w g√≥rńô wewnńÖtrzkom√≥rkowe biaŇāka antyapoptotyczne, w tym biaŇāko FLIP, surwiwinńô, cIAP-2, A1/Bfl-1, XIAP (43) i zwińôksza aktywnoŇõńá telomerazy (Ryc. 2) (44) Inhibitory receptora IGF-1 (IGF-1R) wykazywaŇāy w badaniach przedklinicznych aktywnoŇõńá przeciw-MM, (45) dostarczajńÖc podstaw do wykorzystania w przyszŇāych protokoŇāach klinicznych.
Czynnik wywodzńÖcy sińô z kom√≥rek zrńôbowych-1őĪ (SDF-1őĪ)
SDF-1őĪ, poŇõredniczńÖcy w migracji prawidŇāowych hematopoetycznych kom√≥rek zrńôbowych. Obecny jest w osoczu oraz w nadsńÖczach uzyskanych z kom√≥rek podŇõcieliska szpiku kostnego uzyskanych od pacjent√≥w z MM. (46) Receptor CXCR4 dla SDF-1őĪ wykazuje ekspresjńô na niekt√≥rych kom√≥rkach MM, a SDF-1őĪ aktywuje MAPK, PI3K/Akt i NFőļB w kom√≥rkach MM, z nastńôpowńÖ proliferacjńÖ, migracjńÖ oraz ochronńÖ przeciwko apoptozie indukowanej przez dexametazon (Ryc. 2). Na obszarze mikroŇõrodowiska szpiku kostnego, SDF-1őĪ reguluje zwińôkszone wydzielanie IL-6 i VEGF w szpiku kostnym, tym samym dalej wspierajńÖc wzrost, przeŇľycie, opornoŇõńá na leki i migracjńô kom√≥rek nowotworowych (Ryc. 1). Efekty dziaŇāania SDF-1őĪ sńÖ jednak niewielkie i nie stanowi on obiecujńÖcego celu nowatorskich sposob√≥w leczenia MM.
Inne cytokiny
Rekombinowana IL-1ő≤ pobudza kom√≥rki MM do produkcji IL-6, w ten spos√≥b sprzyjajńÖc proliferacji kom√≥rek nowotworowych. (19) TGF-ő≤ wydzielany przez kom√≥rki MM wywoŇāuje wydzielanie IL-6 w podŇõcielisku szpiku i przyczynia sińô do niedobor√≥w immunologicznych charakterystycznych dla MM na drodze zmniejszonej ekspresji limfocyt√≥w B, T i kom√≥rek NK. IL-10 jest czynnikiem proliferacji, lecz nie czynnikiem r√≥Ňľnicowania w przypadku ludzkich kom√≥rek MM. (47) Makrofagowe biaŇāko zapalne-1őĪ jest silnym czynnikiem stymulacji osteoklast√≥w w szpiku kostnym.48 Donoszono o autokrynnym poŇõredniczeniu wzrostu w liniach kom√≥rek MM przez IL-1549 i IL-21. (50) R√≥wnieŇľ cytokiny inne niŇľ IL-6, wykorzystujńÖce sygnalizacjńô przez gp130,w tym onkostatyna M, (51) IL-11 i czynnik hamujńÖcy biaŇāaczkńô ‚Äď1, (52) uznawano za zaangaŇľowane w patogenezie szpiczaka mnogiego.
Nowe leki stosowane w terapii szpiczaka mnogiego
W trakcie oceny znajduje sińô wiele klas nowych lek√≥w, zar√≥wno w badaniach przedklinicznych, jak i klinicznych. Leki te sklasyfikowano wedŇāug miejsc, na kt√≥re dziaŇāajńÖ i zebrano w Tabeli 1.
Leczenie ukierunkowane zar√≥wno na kom√≥rki szpiczaka mnogiego, jak i interakcje kom√≥rek nowotworowych z mikroŇõrodowiskiem szpiku kostnego
Talidomid i jego analogi
Talidomid (Thal) – (alfa-N{ftalimido}glutarimid) jest pochodnńÖ kwasu glutaminowego, klasyfikowanńÖ farmakologicznie jako Ňõrodek immunomodulujńÖcy. (53) Hamuje wytwarzanie TNF-őĪ, (54) a takŇľe blokuje angiogenezńô poprzez inhibicjńô zasadowego czynnika wzrostu fibroblast√≥w (bFGF) i/lub VEGF. (55) Talidomid zostaŇā wycofany z praktyki klinicznej w latach 60-tych XX-tego wieku, z powodu doniesieŇĄ o teratogennoŇõci i licznych przypadk√≥w fokomelii wŇõr√≥d dzieci matek zaŇľywajńÖcych ten lek podczas cińÖŇľy. Jednak niedawno talidomid i jego pochodne (ImiDs) zastosowano w leczeniu chor√≥b zapalnych i nowotwor√≥w zŇāoŇõliwych.
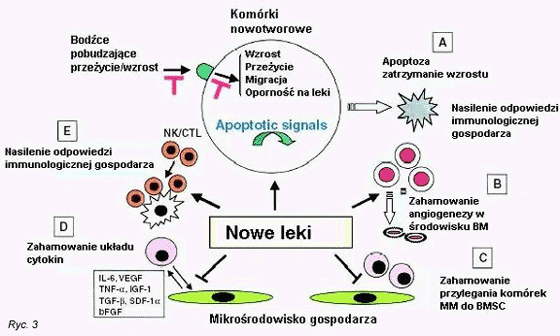
Leki te wykazujńÖ liczne aktywnoŇõci przeciw kom√≥rkom MM. Po pierwsze, dziaŇāajńÖ bezpoŇõrednio na kom√≥rki nowotworowe indukujńÖc zatrzymanie fazy G1 wzrostu lub apaptozńô, nawet w opornych na leki liniach kom√≥rek MM i kom√≥rkach pochodzńÖcych od pacjent√≥w. (56) Talidomid hamuje wywoŇāywanńÖ przez TNF-őĪ i IL-1ő≤ w kom√≥rkach Jurkat aktywnoŇõńá wińÖzania heterodimeru p50/p65 NF-őļB z DNA, (57) a IMiDs hamujńÖ czynnoŇõńá NF-őļB w kom√≥rkach MM. (56) Aktywacja NF-őļB reguluje przeŇľycie kom√≥rek, procesy antyapoptotyczne i wytwarzanie cytokin w szpiczaku mnogim, (15) a zahamowanie aktywacji NF-őļB przez Thal/IMiDs nasilańá moŇľe wraŇľliwoŇõńá na inne Ňõrodki chemioterapeutyczne. Apoptoza kom√≥rek MM indukowana przez Thal/IMiDs poŇõredniczona jest przez kaspazńô-8, co sugeruje potencjalne korzyŇõci poŇāńÖczenia ich z lekami ukierunkowanymi na apoptozńô poŇõredniczonńÖ przez kaspazńô-8 (tj. Dex) w celu wywoŇāania podw√≥jnego sygnaŇāu apoptotycznego. (56,58) Po drugie, Thal/IMiDs hamujńÖ przyleganie kom√≥rek MM do podŇõcieliska szpiku kostnego, pokonujńÖc w ten spos√≥b CAM-DR. (13) Po trzecie, Thal/IMiDs hamujńÖ w kom√≥rkach MM i/lub podŇõcielisku szpiku kostnego aktywnoŇõńá biologicznńÖ, wydzielanie cytokin zwińôkszajńÖcych wzrost, przeŇľycie, opornoŇõńá na leki i migracjńô kom√≥rek MM. Po czwarte, Ňõrodki te wykazujńÖ aktywnoŇõńá antyangiogennńÖ, poŇõrednio na drodze hamowania bFGF i/lub VEGF. (55) Po pińÖte, Thal/IMiDs mogńÖ wywierańá dziaŇāanie przeciwko MM poprzez efekty immunomodulujńÖce, takie jak indukcja odpowiedzi limfocyt√≥w pomocniczych Th1 z wydzielaniem interferonu-ő≥ (IFN-ő≥) i IL-2. (59) Wykazano, Ňľe w szpiczaku mnogim Thal/IMiDs indukowańá mogńÖ, zar√≥wno in vitro, jak i in vivo przeciwnowotworowńÖ odpornoŇõńá kom√≥rkowńÖ limfocyt√≥w NK (Ryc. 3). (60)
Od kilku lat, pod przewodnictwem prof. A. DmoszyŇĄskiej, sńÖ prowadzone w Polsce badania kliniczne zwińÖzane z wykorzystaniem talidomidu u chorych z MM. Wyniki badaŇĄ zaprezentowane w czasie IX Mińôdzynarodowych Warsztat√≥w Naukowych dotyczńÖcych MM w Salamance (61), u ponad 175 chorych z nawrotowńÖ lub pierwotnie opornńÖ postacińÖ MM, wykazaŇāy iŇľ odsetek odpowiedzi na leczenie wynosiŇā 56%. W grupie pacjent√≥w odpowiadajńÖcych na talidomid zaznaczyŇāa sińô tendencja do szybkiej normalizacji stńôŇľenia biaŇāka caŇākowitego, czńôsto juŇľ po 4 tygodniach od rozpoczńôcia terapii oraz powolnego obniŇľania stńôŇľenia biaŇāka M, poŇāńÖczonej z poprawńÖ parametr√≥w aktywnoŇõci choroby typowych dla MM, takich, jak plazmocytoza szpiku kostnego, beta2-mikroglobulina i aktywnoŇõńá dehydrogenazy mleczanowej. W badaniach polskich potwierdzono, iŇľ talidomid hamuje sekrecjńô cytokin prozaplanych i proangiogennych, takich jak: IL-6, TNFa, VEGF, b-FGF, HGF u chorych na MM63 Wykazano r√≥wnieŇľ, iŇľ lek ten ma wpŇāyw immunomodulujńÖcy na sekrecjńô cytokin wydzielanych przez limfocyty T (IL-1b, IL-6, IL-2, IFN g, zwińôksza wytwarzanie IL-4, IL-5, IL-8 oraz hamuje wytwarzanie IL-12 i TNFa64.
W badaniu klinicznym II fazy dotyczńÖcym leczenia talidomidem u chorych z nawrotowym i opornym na leczenie MM wykazano odpowiedŇļ pozytywnńÖ u 32 % pacjent√≥w, co wińÖzaŇāo sińô z trwale zmniejszonym odsetkiem kom√≥rek plazmatycznych w szpiku kostnym i zwińôkszonymi stńôŇľeniami hemoglobiny. (62) W innych badaniach klinicznych dotyczńÖcych leczenia talidomidem u pacjent√≥w z MM opornym na konwencjonalne sposoby leczenia uzyskano odpowiedzi u okoŇāo 25 % pacjent√≥w. (65) W dw√≥ch innych badaniach wykorzystujńÖcych poŇāńÖczenie Dex i Thal uzyskano odpowiedzi u okoŇāo dw√≥ch trzecich pacjent√≥w ze ŇõwieŇľo rozpoznanym MM, bez toksycznego naraŇľania nastńôpnie pozyskiwanych autologicznych kom√≥rek macierzystych z krwi obwodowej. (66) Zatem poŇāńÖczenie talidomidu z dexametazonem reprezentuje nowatorski skuteczny schemat leczenia.
ZakoŇĄczono badanie fazy I nad leczeniem Revimidem (IMiD3, CC-5013) u pacjent√≥w z nawrotowym i opornym na leczenie MM. (67) W tym badaniu u pacjent√≥w z postńôpujńÖcym MM i narastajńÖcńÖ paraproteinemińÖ pomimo leczenia konwencjonalnego, u dw√≥ch trzecich os√≥b uzyskano godnńÖ uwagi aktywnoŇõńá klinicznńÖ przeciw-MM, udowodnionńÖ poprzez 25 % zmniejszenie biaŇāka monoklonalnego w surowicy, a ustabilizowanie sińô lub zmniejszenie paraprotein MM widoczne byŇāo u 79 % pacjent√≥w. Co istotne, nie obserwowano sennoŇõci, neuropatii i zaparńá zwińÖzanych z leczeniem talidomidem. WcińÖŇľ trwajńÖce badanie fazy II nad Revimidem ujawniŇāo stabilizacjńô choroby lub pozytywnńÖ odpowiedŇļ (w tym kilka caŇākowitych remisji) u 39 z 46 pacjent√≥w z chorobńÖ nawrotowńÖ lub opornńÖ na leczenie. W oparciu o tńÖ obiecujńÖcńÖ aktywnoŇõńá anty-nowotworowńÖ i bardzo korzystny profil skutk√≥w ubocznych, niedŇāugo rozpocznńÖ sińô badania fazy II w celu okreŇõlenia uŇľytecznoŇõci klinicznej Revimidu u pacjent√≥w ze ŇõwieŇľo rozpoznanym MM, w momencie pierwszego nawrotu i jako leczenia podtrzymujńÖcego po wykonaniu autoprzeszczepu.
Inhibitor proteasomów PS-341
WewnńÖtrzkom√≥rkowy, biochemiczny szlak ubikwityny/proteasom√≥w jest systemem proteolitycznym zar√≥wno w cytozolu, jak i w jńÖdrze, regulujńÖcym cykliny oraz kinazowe inhibitory biaŇākowe zaleŇľne od cyklin, tym samym regulujńÖc postńôp cyklu kom√≥rkowego. (68) PS-341 (VelcadeTM) reprezentuje klasńô inhibitor√≥w proteasom√≥w ‚Äď boronian√≥w peptydowych, hamujńÖcych aktywnoŇõńá proteasom√≥w 26S. (69) PoczńÖtkowym uzasadnieniem dla zastosowania inhibitor√≥w proteasom√≥w w MM byŇāo ich hamujńÖce dziaŇāanie na aktywacjńô NF-őļB. Reguluje on zar√≥wno transkrypcjńô IL-6 w kom√≥rkach podŇõcieliska szpiku kostnego, ekspresjńô czńÖsteczek adhezyjnych (CD54 i CD106) na kom√≥rkach MM i zrńôbu, jak i ekspresjńô biaŇāek cyklu kom√≥rkowego (cyklina D1) i antyapoptotycznych (IAPs, Bcl-xL) w kom√≥rkach nowotworowych.
Aktywacja NF-őļB nadaje kom√≥rkom MM opornoŇõńá na leki, reguluje ekspresjńô czńÖsteczek adhezyjnych na patologicznych plazmocytach i kom√≥rkach podŇõcieliska szpiku kostnego. Ponadto reguluje zar√≥wno konstytutywnńÖ, jak i zaleŇľnńÖ od przylegania kom√≥rek MM transkrypcjńô oraz wydzielanie cytokin. (14,15) PS-341 blokuje degradacjńô IőļBőĪ (biaŇāka inhibitorowego zwińÖzanego z cytozolowym NF-őļB), hamujńÖc w ten spos√≥b IőļBőĪ oraz zwińÖzane z tym translokacje jńÖdrowe i wińÖzanie NF-őļB ze swoim zgodnym motywem wińÖzania w DNA. W zwińÖzku z tym PS-341 jest nowym Ňõrodkiem terapeutycznym dla pokonania opornoŇõci na leki w kom√≥rkach MM, hamuje wińÖzanie patologicznych plazmocyt√≥w w szpiku kostnym i wydzielanie cytokin. Badania in vitro potwierdziŇāy, Ňľe PS-341 indukuje apoptozńô poprzez aktywacje kaspazy -8, -9 i ‚Äď3 w opornych na leki liniach kom√≥rkowych MM oraz u pacjent√≥w ze szpiczakiem mnogim. (70) PS-341 reguluje ekspresjńô czńÖsteczek adhezyjnych na kom√≥rkach MM oraz podŇõcielisku szpiku kostnego i ŇāńÖczńÖce sińô z tym wińÖzanie. (15) Blokuje konstytutywne i indukowane przez przyleganie kom√≥rek MM, zaleŇľne od NF-őļB wydzielanie cytokin w podŇõcielisku szpiku oraz hamuje angiogenezńô. (71)
ChociaŇľ PS-341 hamuje aktywacjńô NF-őļB, to molekularne mechanizmy jego aktywnoŇõci przeciw-MM nie sńÖ w peŇāni okreŇõlone. Profil mikromacierzy genowych wykazuje, Ňľe PS-341 reguluje transkrypcjńô gen√≥w wzrostu i przeŇľycia ŇāńÖcznie z indukcjńÖ trankrypt√≥w gen√≥w apoptotycznych, ubikwityny/proteasom√≥w i odpowiedzi na stres w kom√≥rkach MM. Badania te okreŇõlajńÖ czuŇāoŇõńá dotyczńÖcńÖ cel√≥w molekularnych i opornoŇõńá na PS-341, dostarczajńÖ uzasadnienia dla strategii ŇāńÖczenia z lekami konwencjonalnymi lub nowymi oraz uwzglńôdniajńÖ rozw√≥j celowanych lek√≥w nastńôpnej generacji, silnych, wybi√≥rczych i mniej toksycznych. Przeprowadzone badania wykazaŇāy, Ňľe PS-341 hamuje naprawńô DNA poprzez cińôcie podjednostki katalitycznej zaleŇľnej od DNA kinazy biaŇākowej. (70) Leczenie linii kom√≥rkowych MM opornych na Ňõrodki uszkadzajńÖce DNA (melfalan, antracyklina) tymi lekami, na kt√≥re sńÖ one oporne, z nastńôpowym (po 12-24 godz) podawaniem subletalnych dawek PS-341, moŇľe hamowańá naprawńô uszkodzeŇĄ DNA i przywr√≥cińá wraŇľliwoŇõńá na leki. (72) PS-341 indukuje takŇľe zaleŇľne od kaspaz cińôcie gp130 (CD130), ő≤-podjednostki IL-6R,73 tym samym blokujńÖc poŇõredniczone przez IL-6 dalsze przekaŇļnictwo na szlakach sygnaŇāowych Raf/MEK/a42/44 MAPK, JAK2/STAT3 PI3K/Akt poŇõredniczńÖcych odpowiednio wzroŇõcie, przeŇľyciu i opornoŇõci na leki. BiorńÖc pod uwagńô znaczenie sygnalizacji przez gp130 dla IL-6 i cytokin pokrewnych, ta wywoŇāywana przez PS-341 degradacja gp130 odpowiadańá moŇľe, przynajmniej po czńôŇõci, za jego nadzwyczajnńÖ aktywnoŇõńá anty-nowotworowńÖ. W wielooŇõrodkowym badaniu II fazy nad zastosowaniem PS-341 u pacjent√≥w z nawrotowym, opornym na leczenie MM osińÖgnińôto okoŇāo 35 % odpowiedzi, w tym wiele trwaŇāych odpowiedzi caŇākowitych (mediana 12 miesińôcy). Pozytywne odpowiedzi wińÖzaŇāy sińô z korzyŇõciami klinicznymi, w tym poprawńÖ jakoŇõci Ňľycia, zwińôkszonym stńôŇľeniem hemoglobiny i zmniejszeniem iloŇõci transfuzji preparat√≥w krwiopochodnych, poprawńÖ czynnoŇõci nerek oraz zwińôkszeniem stńôŇľeŇĄ prawidŇāowych immunoglobulin. (74) W wińôkszoŇõci przypadk√≥w moŇľna byŇāo Ňāatwo poradzińá sobie z dziaŇāaniem ubocznym leku tj. zmńôczeniem i dolegliwoŇõciami ŇľoŇāńÖdkowo-jelitowymi. Trombocytopenia i neuropatia wystńôpowaŇāy gŇā√≥wnie u pacjent√≥w, u kt√≥rych te stany chorobowe juŇľ istniaŇāy uprzednio. Aktualnie PS-341 oceniane jest w badaniach fazy II u pacjent√≥w z MM we wczeŇõniejszych stadiach (ŇõwieŇľo rozpoznanym, przy pierwszym nawrocie) i jest por√≥wnywany z dexametazonem w mińôdzynarodowym, wielooŇõrodkowym badaniu fazy III u pacjent√≥w z nawrotem MM.
Trójtlenek arsenu
Tr√≥jtlenek arsenu (As203) r√≥wnieŇľ wywoŇāuje apoptozńô na drodze aktywacji kaspazy-9 w liniach kom√≥rek MM opornych na leczenie i patologicznych plazmocytach pochodzńÖcych od pacjent√≥w. Hamuje r√≥wnieŇľ aktywacjńô JAK/STAT3 i zwińôksza Mcl-1 wywoŇāywanńÖ przez IL-6 oraz hamuje aktywacjńô NFőļB w podŇõcielisku szpiku kostnego. W ten spos√≥b obniŇľa ekspresjńô CD54 i wińÖzanie kom√≥rka nowotworowa- kom√≥rka zrńôbu, jak i hamuje transkrypcjńô oraz parakrynne wydzielanie IL-6 i VEGF.75 Lek ten zwińôksza niszczenie kom√≥rek MM poŇõredniczone przez limfocyty LAK poprzez selektywne zwińôksznie ekspresji CD38 i CD54 na kom√≥rkach MM, jak r√≥wnieŇľ CD31 i CD11a na kom√≥rkach LAK. (76) W badaniach klinicznych fazy I/II nad stosowaniem As203 u pacjent√≥w z MM opornym na leki i nawrotowym wykazano nieznaczne zmniejszenia lub stabilizacjńô paraproteiny MM. Skutki uboczne zastosowania As203 obejmujńÖ leukopenińô, niedokrwistoŇõńá, b√≥le brzucha, biegunkńô, gorńÖczkńô oraz zmńôczenie. (77) Ponadto, indukowanńÖ As203 cytotoksycznoŇõńá wzglńôdem kom√≥rek szpiczakowych moŇľe nasilańá kwas askorbinowy. (78) R√≥wnieŇľ dexametazon nasila in vitro apoptozńô indukowanńÖ przez As203, (75) dostarczajńÖc uzasadnienia dla badania klinicznego z zastosowaniem tego poŇāńÖczenia.
2-metoksyestradiol
2-metoksyestradiol (2ME2) jest naturalnym metabolitem estradiolu o silnej aktywnoŇõci przeciwnowotworowej i przeciwangiogennej w biaŇāaczce w badaniach in vitro i in vivo. (79) WińÖŇľe sińô sŇāabo z receptorem estrogenowym i poŇõredniczy swoje dziaŇāania przeciwnowotworowe niezaleŇľnie od ekspresji receptora estrogenowego i jego aktywnoŇõci. Ňörodek ten hamuje wzrost i indukuje apoptozńô w liniach kom√≥rek MM opornych na leki. Nasila apoptozńô indukowanńÖ przez deksametazon, przezwycińôŇľa ochronne dziaŇāanie IL-6, IGF-1, jak i zmniejsza wywoŇāane wińÖzaniem kom√≥rek nowotworowych wydzielanie VEGF oraz IL-6 przez podŇõcielisko szpiku kostnego.80 2ME2 indukuje apoptozńô kom√≥rek MM poprzez uruchomienie mitochondrialnego cytochromu c i Smac, z nastńôpowńÖ aktywacjńÖ kaspazy-8, -9 i ‚Äď3. Mechanizmy molekularne przeciwszpiczakowej aktywnoŇõci 2ME2 okreŇõlono z zastosowaniem profilu mikromacierzy genetycznych.33 Obecnie trwajńÖ badania kliniczne fazy II w MM nad wykorzystaniem 2ME2.
Inhibitor acylotransferazy-ő≤ kwasu lizofosfatydowego
Kwas lizofosfatydowy (LPA) i kwas fosfatydowy (PA) sńÖ fosfolipidami zaangaŇľowanymi w przekaŇļnictwo sygnaŇāowe i biosyntezńô lipid√≥w. Swoiste dla izoformy czynnoŇõciowe inhibitory enzymu acylotransferazy LPA, konwertujńÖcej LPA do PA, wykazujńÖ silnńÖ cytotoksycznoŇõńá wzglńôdem linii kom√≥rkowych MM i kom√≥rek szpiku pacjent√≥w. Inhibitory LPAAT-ő≤ wywoŇāujńÖ apoptozńô w kom√≥rkach MM poprzez uszkodzenie polimerazy poli-ADP-rybozy (PARP), nawet przy obecnoŇõci IL-6, IGF-1 i kom√≥rek podŇõcieliska (Ryc. 3) (81) dostarczajńÖc podstawy dla badaŇĄ klinicznych.
Nowe leki ukierunkowane na szlaki zwińÖzane z poŇõredniczeniem wzrostu i przeŇľycia kom√≥rek szpiczaka mnogiego
PTK787/ZK222584
Kilku badaczy opisaŇāo istotnńÖ rolńô VEGF w MM. (17,82) VEGF wydzielany jest przez kom√≥rki szpiczakowe i kom√≥rki podŇõcieliska szpiku kostnego oraz zwińôksza wydzielanie IL-6 przez zrńÖb szpiku kostnego. (16,21,82) VEGF wywoŇāuje fosforylacjńô Flt-1 i dalszńÖ aktywacjńô p42/44 MAPK oraz proliferacjńô, hamowane przez przeciwciaŇāa przeciw-VEGF lub inhibitor MEK PD98059.17 VEGF indukuje migracjńô kom√≥rek MM, blokowanńÖ przez inhibitor PKC chlorowodorek bisindolylomaleimidu I (Ryc. 2). VEGF wywoŇāuje takŇľe aktywacjńô PI3K/Akt. Wsp√≥lnie z poŇõredniczonym prze ő≤1-integrynńô wińÖzaniem fibronektyny, PI3K wywoŇāuje rekrutacjńô i aktywacjńô PKCőĪ, tym samym nasilajńÖc migracjńô kom√≥rek MM. (33) Inhibitor kinazy tyrozynowej VEGFR PTK787/ZK222584 blokuje indukowanńÖ przez VEGF fosforylacjńô tyrozyn w Flt-1, aktywacjńô MEK/MAPK i proliferacjńô. Blokuje on takŇľe migracjńô zaleŇľnńÖ od aktywacji PKC (Ryc. 3), (37) co stwarza podstawńô dla badania klinicznego nad PTK787/ZK222584 u chorych z nawrotowym MM.
Inhibitory transferazy farnezylowej
Cytozolowy enzym transferaza farnezylowa przenosi grupy farnezylowe z dwufosforanu farnezylu na motyw CAAX biaŇāka Ras, uŇāatwiajńÖc tym samym jego przyczepienie do bŇāony wewnńôtrznej kom√≥rki plazmatycznej i wińÖŇľńÖce sińô z tym przekaŇļnictwo sygnaŇāowe.83 Hamowanie farnezylacji jest strategińÖ blokowania aktywnoŇõci Ras, a kilka inhibitor√≥w transferazy farnezylowej (FTIs) hamuje wzrost kom√≥rek nowotworowych zar√≥wno w badaniu in vitro, jak i in vivo.84 Indukowana przez cytokiny (IL-6, VEGF i IGF-1) proliferacja kom√≥rek MM poŇõredniczona jest przez sygnalizacjńô Ras/Raf/MAPK (Ryc. 2 i 3), (17,43) co dostarcza przedklinicznego uzasadnienia dla trwajńÖcych badaŇĄ klinicznych fazy I/II nad zastosowaniem w MM dw√≥ch FTIs, SCH-6633685 i R115777. (86)
Inhibitory deacetylazy histonów
Acetylacja histon√≥w moduluje ekspresjńô gen√≥w, r√≥Ňľnicowanie i przeŇľycie kom√≥rek. Jest ona regulowana przez acetylotransferazy histon√≥w i deacetylazy histon√≥w (HDACs). Nowe oparte na kwasie hydroksamowym hybrydowe zwińÖzki chemiczne, takie jak SAHA, sńÖ inhibitorami deacetylazy histon√≥w. IndukujńÖ gromadzenie sińô acetylowanych nukleosomalnych rdzeni histonowych z wińÖŇľńÖcńÖ sińô z tym indukcjńÖ r√≥Ňľnicowania i/lub apoptozy w kom√≥rkach transformowanych i nowotworowych. SAHA indukuje zatrzymanie wzrostu i apoptozńô w kom√≥rkach pacjent√≥w z MM oraz szpiczakowych liniach kom√≥rkowych, nawet tych opornych na Dex i leczenie konwencjonalne.87 Inhibitor HDAC LAQ824, kwas cinnamylowo-hydroksamowy, indukuje zaleŇľnńÖ od kaspaz apoptozńô kom√≥rek MM oraz hamuje zar√≥wno aktywnoŇõńá proteasom√≥w 20S, jak i konstytutywnńÖ aktywacjńô NF-őļB w MM (Ryc. 3). (88)
Inhibitory biaŇāka szoku termicznego-90
BiaŇāko szoku termicznego-90 uŇāatwia wewnńÖtrzkom√≥rkowe przemieszczanie, dojrzewanie konformacyjne i faŇādowanie tr√≥jwymiarowe wymagane dla czynnoŇõci biaŇāka. Antybiotyk ansamycynowy geldanamycyna (GA) i jej analogi wińÖŇľńÖ sińô z krytycznym miejscem wińÖzania adenozynotr√≥jfosforanu w Hsp90, w ten spos√≥b znoszńÖc jego aktywnoŇõńá w Ňõrodowisku szpiku kostnego, zmniejszajńÖc ekspresjńô IGF-1R i IL-6R na kom√≥rkach MM, powodujńÖc deplecjńô kinaz wzrostowych [np. Akt, kinazy IőļB (IKK) i Raf] oraz biaŇāek przeciwapoptotycznych (FLIP, XIAP, cIAP i telomerazy), jak i hamujńÖc indukowanńÖ przez cytokiny aktywacjńô NF-őļB i telomerazy (hTERT). (89) GA oraz inne inhibitory Hsp90 indukujńÖ apoptozńô szpiczakowych linii kom√≥rkowych i kom√≥rek pacjent√≥w z MM, opornych na Dex, antracykliny, Thal lub ImiDs i PS-341. Profil mikromacierzy genowych wykazuje, Ňľe PS-341 indukuje Hsp90 w kom√≥rkach MM. Przeciwnie, blokowanie tej odpowiedzi przez GA nasila wywoŇāanńÖ przez PS-341 apoptozńô kom√≥rki. GA wykazuje aktywnoŇõńá przeciw-MM w modelu ludzkiego szpiczaka u myszy ze SCID, i bńôdzie ona w przyszŇāoŇõci oceniana w protokoŇāach klinicznych zar√≥wno sama, jak i ŇāńÖcznie z PS-341.
GenasenseTM (G3139, oligonukleotyd antysensowny Bcl-2)
CzŇāonkowie rodziny biaŇāek Bcl-2 (tj. Bcl-2, Bcl-xL, Bcl-xs i Bax) regulujńÖ apoptozńô i opornoŇõńá na leki w MM. Nadmierna ekspresja Bcl-2 korelowaŇāa r√≥wnieŇľ z opornoŇõcińÖ na IFN. (90) Apoptoza indukowana przez dexametazon w szpiczakowych liniach kom√≥rkowych ARP-1 i RPMI 8226 kom√≥rek MM koreluje z poziomem ekspresji Bcl-2. ŇöciŇõle rzecz biorńÖc opornoŇõńá na dexametazon, ale nie na melfalan, wińÖŇľe sińô z nadmiernńÖ ekspresjńÖ Bcl-2.91 GenasenseTM jest oligonukleotydem antysensownym Bcl-2, zmniejszajńÖcym stńôŇľenia mRNA i biaŇāka Bcl-2 w szpiczakowych liniach kom√≥rkowych MM i kom√≥rkach MM pacjent√≥w. (92) W badaniach fazy III por√≥wnuje sińô zastosowanie w szpiczaku mnogim Dex vs Dex z GenasenseTM.
Nowe leki ukierunkowane na interakcje kom√≥rek nowotworowych z mikroŇõrodowiskiem szpiku kostnego
Inhibitor kinazy IőļB PS-1145
Nowy swoisty inhibitor IKK PS-1145 tylko czńôŇõciowo hamuje proliferacjńô kom√≥rek MM, co sugeruje, Ňľe blokada NF-őļB nie moŇľe odpowiadańá za caŇāńÖ aktywnoŇõńá przeciwnowotworowńÖ inhibitora proteasom√≥w PS-341. PS-1145 blokuje aktywacjńô NF-őļB w kom√≥rkach MM i podŇõcieliska szpiku kostnego. Blokuje takŇľe dalsze nastńôpstwa, w tym ekspresjńô czńÖsteczek adhezyjnych i wińÖzanie kom√≥rka MM-podŇõcielisko, proliferacjńô przylegajńÖcych kom√≥rek MM, jak r√≥wnieŇľ transkrypcjńô i wydzielanie IL-6 (Ryc. 2). Badania te zwracajńÖ uwagńô na znaczenie dziaŇāania nowych Ňõrodk√≥w terapeutycznych na patologiczne plazmocyty w ich mikroŇõrodowisku szpiku kostnego.
Inhibitor p39 MAPK
BiaŇāko p38 MAPK aktywowane jest przez cytokiny i czynniki wzrostu. Aktywacja p38 MAPK wińÖŇľe sińô z ekspresjńÖ genu i/lub wydzielaniem IL-6 w kom√≥rkach Sertoli‚Äôego, (93) kom√≥rkach mińôŇõnia sercowego (94) i osoteoblastach. (95) Swoisty inhibitor p38 MAPK VX-745 hamuje wydzielanie IL-6 i VEGF w podŇõcielisku szpiku kostnego, jak r√≥wnieŇľ indukowane przez TNF-őĪ wydzielanie IL-6. VX-745 hamuje takŇľe zar√≥wno proliferacjńô kom√≥rek MM, jak i wydzielanie IL-6 w mikroŇõrodowisku szpiku kostnego spowodowane przyleganiem patologicznych plazmocyt√≥w do zrńôbu (Ryc. 3). (22) Badania te rozpoznajńÖ w p38 MAPK nowy cel terapeutyczny, kt√≥ry byńá moŇľe jest warty wykorzystania, aby przezwycińôŇľyńá opornoŇõńá na dotychczasowe leczenie i poprawińá wyniki terapeutyczne u pacjent√≥w z MM.
Nowe leki ukierunkowane na receptory powierzchni komórki
IndukujńÖcy apaoptozńô ligand pokrewny czynnikowi martwicy nowotwor√≥w/ligand Apo2
TRAIL/Apo2L (czŇāonek nadrodziny TNF ‚Äď ligand√≥w indukujńÖcych Ňõmierńá, obejmujńÖcej TNF-őĪ i ligand Fas), indukuje poŇõredniczonńÖ przez kaspazńô-8 apoptozńô ludzkich linii kom√≥rkowych MM i kom√≥rek pochodzńÖcych ze szpiku kostnego pacjent√≥w, w tym tych, kt√≥re sńÖ oporne na konwencjonalne sposoby leczenia. (96) TRAIL/Apo2L hamuje takŇľe wzrost ludzkich kom√≥rek MM u myszy ze SCID. (96) Dlatego teŇľ, leki indukujńÖce sygnalizacjńô apoptotycznńÖ TRAIL mogńÖ przezwycińôŇľyńá klinicznńÖ opornoŇõńá na leki w MM.
Inhibitory receptora insulinopodobnego czynnika wzsrostu-1
Inhibitor kinazy tyrozynowej IGF-1R ADW poŇõredniczy w aktywnoŇõci przeciwnowotworowej zar√≥wno w badaniach in vitro, jak i w modelu ludzkiego MM u myszy z cukrzycńÖ i ze SCID, a bez otyŇāoŇõci. (45) PoniewaŇľ IGF-1 jest waŇľnym czynnikiem przeŇľycia w MM (Ryc. 2), (28,43,97) to blokowanie kaskad sygnaŇāowych poŇõredniczonych przez IGF-1 stanowi obiecujńÖcńÖ opcjńô terapeutycznńÖ.
Statyny
Statyny (lowastatyna i fluwastatyna) sńÖ nieodwracalnymi inhibitormi redukatazy ő≤-hydroksy-ő≤-metyloglutarylo-koenzymu A, co blokuje wytwarzanie mawelonianu, krytycznego skŇāadnika w syntezie cholesterolu i izoprenoid√≥w. Prenylacja biaŇāek docelowych, w tym biaŇāka Ras wińÖŇľńÖcego guanozyno-tr√≥jfosforan, jest niezbńôdna dla ich umiejscowienia bŇāonowego i sygnalizacji wewnńÖtrzkom√≥rkowej. Lowastatyna skutecznie indukuje apoptozńô w liniach kom√≥rkowych MM i jest skuteczna w poŇāńÖczeniu z innymi lekami. (98) Na przykŇāad, lowastatyna indukuje apoptozńô w kom√≥rkach MM pacjent√≥w, nawet tych opornych zar√≥wno na konwencjonalne, jak i nowe (IMiDs, PS-341) Ňõrodki terapeutyczne. (99)
PrzeciwciaŇāo przeciw-CD20 (rituksymab)
Terapia skierowana przeciwko CD20 obejmuje jedynie mniejszoŇõńá kom√≥rek nowotworowych MM pacjent√≥w, poniewaŇľ ekspresja CD20 w MM nie jest zbyt czńôsta (20 % CD20+). Leczenie przeciwciaŇāem monoklonalnym anty-CD20, rituksymabem, daŇāo odpowiedŇļ u okoŇāo 32 % uprzednio leczonych pacjent√≥w z MM, gdzie wszyscy mieli CD20+ kom√≥rki nowotworowe. (100)
Immunoterapia oparta na komórkach dendrytycznych
Kom√≥rki dendrytyczne (DCs) odgrywajńÖ centralnńÖ rolńô w prezentacji antygenu (Ag) naiwnym limfocytom T oraz w indukcji pierwotnych odpowiedzi immunologicznych. ChociaŇľ w badaniach przedklinicznych wykazano, Ňľe DCs obŇāadowane Ag ex vivo indukujńÖ silne odpowiedzi przeciwnowotworowe in vitro oraz in vivo w przypadku innych nowotwor√≥w zŇāoŇõliwych, to w MM immunoterapia oparta na kom√≥rkach dendrytycznych nie stanowi na razie uznanej opcji. ChociaŇľ w kilku badaniach wykazano, Ňľe DCs pochodzńÖce od pacjent√≥w z MM mogńÖ wykazywańá prawidŇāowńÖ czynnoŇõńá in vitro, (101) to inne doniesienia wykazujńÖ zmniejszonńÖ reaktywnoŇõńá DC na aktywacjńô CD40. (102) Dhodapkar i wsp. (103) donieŇõli, Ňľe limfocyty T indukowane przez wstrzyknińôte DCs atakujńÖ ŇõwieŇľe autologiczne kom√≥rki nowotworowe, a nie prawidŇāowe kom√≥rki szpiku kostnego. Z podobnńÖ wydajnoŇõcińÖ indukowano limfocyty efektorowe CD8+ pochodzńÖce z krwi obwodowej pacjenta, jak i szpik kostny.
ChociaŇľ pr√≥bowano wielu opartych na DC strategii szczepienia (Ryc. 5), to nie jest na razie jasne, jaki antygen jest optymalny i skuteczny dla obŇāadowania DC. Peptydy wińÖŇľńÖce sińô bezpoŇõrednio z czńÖsteczkami ludzkich Ag leukocyt√≥w (HLA) na DCs sńÖ najlepszymi kandydatami wŇõr√≥d antygen√≥w, ale takie sposoby podejŇõcia wykazujńÖ pewne ograniczenia. WynikajńÖ one z powodu zar√≥wno restrykcji HLA pacjent√≥w, jak i wybi√≥rczoŇõci ekspresji Ag na kom√≥rkach MM. Swoista idiotypowo determinanta regionu zmiennego Ig stanowi idealny Ag docelowy dla przezwycińôŇľenia tych ograniczeŇĄ, poniewaŇľ wykazuje unikatowńÖ ekspresjńô na kom√≥rkach nowotworowego klonu limfocyt√≥w B. Po pomyŇõlnym klinicznie poczńÖtkowym doniesieniu w chŇāoniaku grudkowym, (104) przeprowadzono badania nad szczepieniem DCs obŇāadowanych biaŇākami idiotypowymi u chorych z MM. (105) ChociaŇľ w tych badaniach swoiste idiotypowo limfocyty T cytotoksyczne (CTLs) podlegaŇāy indukcji, to do chwili obecnej nie wykazano by wińÖzaŇāo sińô to z trwaŇāymi odpowiedziami klinicznymi. Sposoby podejŇõcia oparte na stosowaniu pojedynczego Ag wykazujńÖ ograniczenia z powodu odpowiedzi krzyŇľowej z prawidŇāowymi kom√≥rkami lub w zwińÖzku z ewolucjńÖ i ‚ÄěucieczkńÖ‚ÄĚ Ag-ujemnych klon√≥w kom√≥rek nowotworowych. (106)
Inna oparta na DCs strategia szczepienia obejmuje uzyskanie fuzji kom√≥rek MM i DCs z zastosowaniem glikolu polietylenowegoi. Takie kom√≥rki fuzyjne (MM/DC) utworzone z dojrzaŇāych DCs i kom√≥rek MM sńÖ w badaniach in vitro silnymi induktorami odpornoŇõci swoistej dla nowotworu.107 Wytworzone DCs, jako kom√≥rki prezentujńÖce antygen i por√≥wnano dojrzaŇāe DCs z wstrzyknińôtymi ciaŇākami apoptotycznymi MM lub z wstrzyknińôtymi lizatami kom√≥rek MM. DCs z wtrzyknińôtymi ciaŇākami apoptotycznymi MM sńÖ znaczńÖco bardziej skutecznymi induktorami CTLs skierowanych przeciwko autologicznym kom√≥rkom MM, niŇľ DCs z wstrzyknińôtymi lizatami kom√≥rek MM, co dostarcza podstaw dla innych badaŇĄ nad szczepieniami. (108)
Wnioski i przyszŇāe kierunki
Terapie konwencjonalne i wysokodozowane sńÖ w stanie nieznacznie poprawińá wyniki leczenia u chorych z MM. Tymi sposobami terapeutycznymi niewielu, o ile ktokolwiek z pacjent√≥w ulega wyleczeniu. Aktualnie doŇõńá dynamicznie rozwija sińô jednak nowy paradygmat w MM. Oparty jest on na zastosowaniu lek√≥w ukierunkowanych nie tylko na kom√≥rki MM, ale takŇľe na interakcje kom√≥rka nowotworowa – podŇõcielisko szpiku kostnego. Te nowe leki, stosowane same lub w poŇāńÖczeniach z konwencjonalnymi sposobami terapii, sńÖ obiecujńÖce w aspekcie poprawienia ostatecznych wynik√≥w leczenia pacjent√≥w. Co waŇľne, macierze genowe i analiza proteomu pr√≥bek szpiku kostnego, uzyskanych od pacjent√≥w leczonych wedŇāug protokoŇā√≥w klinicznych wykorzystujńÖcych te nowe leki pozwolńÖ okreŇõlińá mechanizmy molekularne wraŇľliwoŇõci i opornoŇõci na leki kom√≥rek nowotworowych, dostarczajńÖc tym samym podstaw dla rozwoju nowej generacji silniejszych i bardziej wybi√≥rczych, a r√≥wnoczeŇõnie mniej toksycznych ukierunkowanych sposob√≥w leczenia szpiczaka mnogiego.
Rycina 1. Interakcje kom√≥rek szpiczaka mnogiego w Ňõrodowisku szpiku kostnego.
Figure 1. Interaction of multiple myeloma cells and their bone marrow milieu.
Rycina 2. Kaskady sygnaŇāowe zwińÖzane ze wzrostem, przeŇľyciem i migracjńÖ kom√≥rek szpiczaka mnogiego.
Figure 2. Signaling cascades mediating growth, survival and migration in multiple myeloma.
Rycina 3. Nowe sposoby leczenia ukierunkowane na kom√≥rki szpiczaka mnogiego i mikroŇõrodowisko szpiku kostnego.
Figure 3. Novel biologically based therapies targeting multiple myeloma cells and the bone marrow microenvironment.
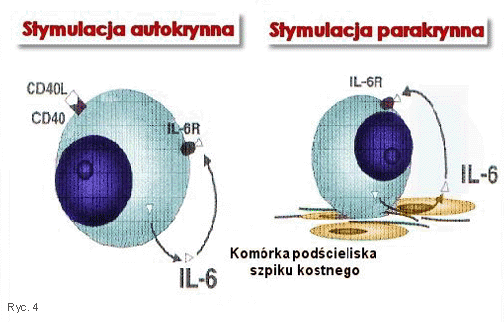
Rycina 4. Schemat autokrynnego i parakrynnego wytwarzania IL-6 przez patologiczne plazmocyty oraz kom√≥rki podŇõcieliska szpiku kostnego u chorego na szpiczaka mnogiego
Figure 4. Draft of autocrine and paracrine IL-6 production by pathologic plasma cells and stroma cells in multiple myeloma patent.
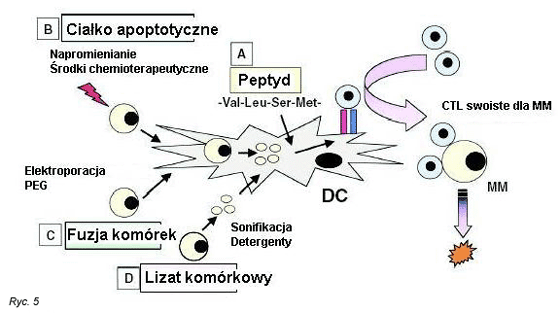
Rycina 5. Strategie oparte na kom√≥rkach dendrytycznych majńÖce na celu wytworzenie limfocyt√≥w T cytotoksycznych przeciw kom√≥rkom szpiczakowym.
Figure 5. Dendritic cells ‚Äď based strategies to generate cytotoxic T lymphocytes against multiple myeloma.
Tabela 1. Nowe leki w terapii szpiczaka mnogiego.
Table 1. New drugs in the therapy of multiple myeloma.
Ukierunkowane zar√≥wno na kom√≥rki MM, jak i interakcje patologicznych plazmocyt√≥w z mikroŇõrodowiskiem szpiku kostnego
|
Ukierunkowane na obwody poŇõredniczńÖce wzrost i przeŇľycie kom√≥rek MM
|
Ukierunkowane na mikroŇõrodowisko szpiku kostnego
|
Ukierunkowane na receptory powierzchni komórek
|
| HMG-CoA ‚Äď redukataza ő≤-hydroksy-ő≤-metyloglutarylo-koenzymu A; IMiDs ‚Äď pochodne immunomodulujńÖce; MAPK ‚Äď aktywowana mitogenami kinaza biaŇākowa; MM ‚Äď szpiczak; S-3APG ‚Äď S-3-[3-amino-ftalimido]-glutarimid; SAHA ‚Äď suberoyanilide hydroxamic acid; TNF ‚Äď czynnik martwicy nowotwor√≥w; VEGF ‚Äď czynnik wzrostu Ňõr√≥dbŇāonka naczyniowego. *W trakcie badaŇĄ klinicznych. |
Publikacja wraz z piŇõmiennictwem dostńôpna w gabinecie dra Artura Jurczyszyna. Zainteresowanych proszńô o kontakt osobisty. Artur Jurczyszyn, Teresa Wolska-SmoleŇĄ, Aleksander B. Skotnicki. Klinika Hematologii, Szpital Uniwersytecki, Krak√≥w.
Katedra i Klinika Hematologii Collegium Medicum Uniwersytetu JagielloŇĄskiego